我國藥品上市許可持有人(MAH)制度的起點可以追溯到2015年����。在此之前,藥品上市許可證是由生產(chǎn)企業(yè)持有和管理的����,這種模式在運行過程中發(fā)現(xiàn)存在一些弊端����,如導致藥品生產(chǎn)同質化現(xiàn)象嚴重����,大量藥企為了拿到生產(chǎn)批件,重復建設生產(chǎn)線����,資源浪費。研發(fā)型企業(yè)����、高校或個人由于無生產(chǎn)場地����,無法注冊申報獲得藥品批件,研發(fā)動力不足等現(xiàn)象����。為了改善藥品行業(yè)整體生態(tài),優(yōu)化資源整合����,鼓勵藥品創(chuàng)新����,中國引入了MAH制度����。2016年6月起的10省市授權試點成果顯著,取得了積極的實施效果����。在試點過程中����,藥品上市許可持有人制度逐漸得到完善和調整,相關政策措施也得到優(yōu)化����。隨著2019年新《藥品管理法》的發(fā)布,藥品上市許可持有人制度正式全國推廣實施����。這為整個藥品行業(yè)帶來了根本性的改變。引入MAH制度以來����,激活了藥品創(chuàng)新研發(fā)的積極性����,提高了藥品上市和市場準入的效率����。
2022年03月31日,國家藥監(jiān)局官網(wǎng)發(fā)布再次公開征求《藥品上市許可持有人檢查要點(征求意見稿)》意見����,MAH文件體系要求企業(yè)建立完善的藥品生產(chǎn)與質量管理、藥品注冊申報和變更管理等制度和文件����,以確保藥品的質量、安全性和合規(guī)性����。這些文件體系有助于提高藥品監(jiān)管的規(guī)范性和有效性,保障公眾的用藥安全����。本文梳理了MAH制度在我國的發(fā)展歷程,參考《藥品上市許可持有人檢查要點(征求意見稿)》等MAH相關法規(guī)制度����,結合本人的理解����,淺析了我國MAH制度文件體系管理要求并整理了制度文件參考清單����,限于個人能力,不足之處還望同仁補充或指正����,以求共同進步。
一����、什么是MAH制度����?
MAH,全稱Marketing Authorization Holder����,中文“上市許可持有人”。簡單來說����,就是持有產(chǎn)品技術的科研個體����、研發(fā)機構����、生產(chǎn)企業(yè)等主體,向行政審批機關提出該產(chǎn)品的上市許可申請����,并依法取得該產(chǎn)品的上市許可批件,承擔產(chǎn)品整個生命周期內質量安全主要責任的制度����。
MAH制度中,持有人和生產(chǎn)單位可以成為同一主體����,也可以是兩個相互獨立的不同主體。上市許可持有人可以根據(jù)自身狀況����,即可以自行生產(chǎn),也可以委托其他有資質的生產(chǎn)企業(yè)進行生產(chǎn)����。在《藥品管理法(2019年版)》中明確指出����,MAH的職責是主要負責藥品全生命周期的質量安全����,應全程監(jiān)督藥品生產(chǎn)過程,且負責產(chǎn)品最終向市場的放行����。
MAH制度下,藥品生產(chǎn)許可證有如下類型����,分別為:
A證:自行生產(chǎn)藥品的上市許可持有人(類似傳統(tǒng)模式下的生產(chǎn)企業(yè));
B證:委托生產(chǎn)藥品的上市許可持有人(新模式下的MAH)����;
C證:受托的生產(chǎn)企業(yè)(無論是否取得A證����,受托生產(chǎn)時必須取得C證)
D證:原料藥生產(chǎn)企業(yè)持有人或代理人(原料藥無法委托生產(chǎn))。
二����、MAH相關法規(guī)制度
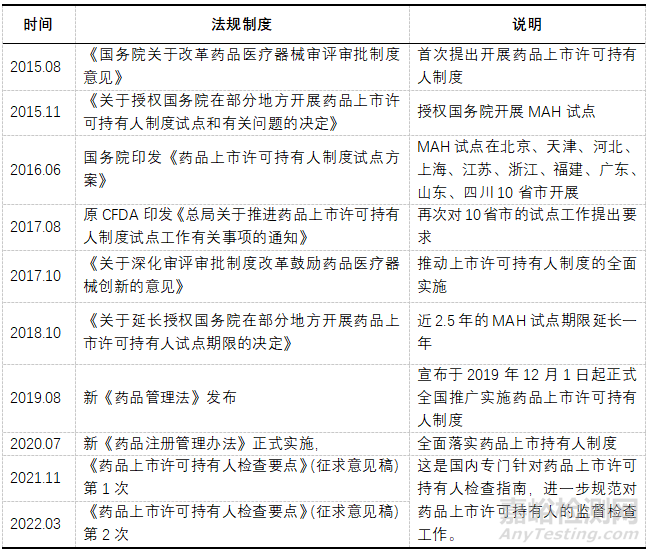
我國藥品上市許可持有人制度是一個從無到有����、不斷完善����、循序漸進的過程,我國自2015年8月《國務院關于改革藥品醫(yī)療器械審評審批制度意見》����,首次提出試點開展藥品上市許可只有人制度開始,直至2019年8月新版《藥品管理法》的實施����,標志著藥品上市許可持有人制度全面落地實施,我國的MAH制度的推廣和施行一直是穩(wěn)步前進����。從法規(guī)制度建設層面,各階段具有里程碑意義的相關事件歸納����,見表1。
▲表1-我國MAH相關法規(guī)制度歷程
三����、MAH文件體系管理要求與參考清單
上市持有人應當建立覆蓋藥品研制����、生產(chǎn)����、銷售、使用全過程的質量保證體系����,持續(xù)強化的質量控制和質量保證能力,依法對藥品研制����、生產(chǎn)、銷售����、使用全過程的安全性、有效性����、質量可控性負責����,人員是關鍵����,硬件是基礎,軟件是保證����。
3.1 MAH文件與記錄
從目前的實際情況來看����,我國許多MAH在硬件建設方面普遍投入充足,人員素質也相對比較高����;但軟件系統(tǒng)的完善不足����,是當前進行質量保證體系的首要任務。2022年3月《藥品上市許可持有人檢查要點(征求意見稿)》對于文件體系有了更具體的要求����,“持有人應當建立保證藥品全生命周期主體責任的規(guī)章制度����。委托其他企業(yè)進行藥品生產(chǎn)����、銷售相關活動(包括藥品儲存����、運輸)的����,相關制度應當與受托企業(yè)的質量管理體系文件有效銜接,并按照規(guī)定形成相關記錄或報告”����,MAH文件與記錄管理����,見表2。
▲表2-MAH文件與記錄管理要求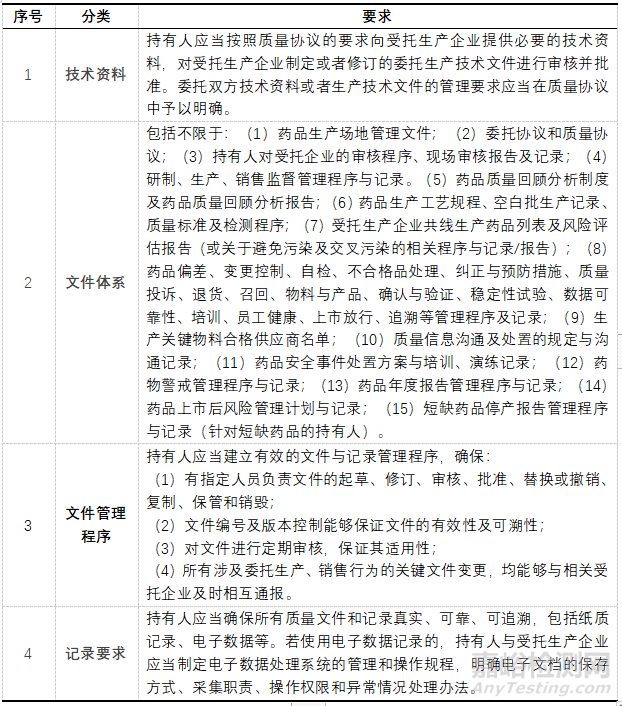
3.2 MAH文件清單
建立一套文件系統(tǒng)成功與否,關鍵是文件總目錄的確定����,先擬定文件目錄草案,這樣可以進行文件編制分工����,明確目標、掌握進度����。在編制過程中不同的MAH主體可以結合自身實際情況對文件目錄進行修改����,文件目錄至少涵蓋崗位職責類文件����、技術標準類文件、管理標準類文件����、操作標準類文件����、記錄類文件等文件目錄。MAH文件體系目錄清單����,見表3����。
▲表3-MAH文件體系目錄清單(供參考)

上述文件清單僅為參考����,不同的MAH公司應以適用和合規(guī)為主要考量指標����,在實際文件體系建設過程中需結合具體情況執(zhí)行����。A證MAH屬于自研自產(chǎn)����,相對來講在文件體系建設方面比較簡單。B證MAH研發(fā)和生產(chǎn)是不同主體����,在文件體系建設方面則較復雜����。如研發(fā)型MAH����,以Biotech+CXO模式為例����,Biotech強項在研發(fā)和創(chuàng)新,而GMP體系下的生產(chǎn)管理(操作����、記錄����、檢驗等)文件體系是短板,故如何建立合規(guī)有效的文件體系����,Biotech與CXO公司雙方需加強溝通����,取長補短����,以利益共同體姿態(tài),在合規(guī)合理的前提下����,制定切實可行的文件管理體系。
四����、結束語
MAH制度要求藥品上市許可持有人對產(chǎn)品的全生命周期負責����。這意味著,在藥品的研發(fā)����、生產(chǎn)和銷售等不同階段����,可能涉及到藥品研發(fā)企業(yè)(持有人)����、受托生產(chǎn)企業(yè)、受托經(jīng)營企業(yè)和受托藥物警戒企業(yè)等各方的工作����。然而,無論工作由誰完成����,MAH制度都要求藥品上市許可持有人與受托方建立起良好的溝通和協(xié)作關系����,確保各方的工作能夠銜接無縫����,并進行有效的監(jiān)督。
作為MAH企業(yè)����,我們不能盲目地照搬GMP各章節(jié)來起草質量體系文件。關鍵在于要對受托方進行審計����,通過審計發(fā)現(xiàn)問題,明確雙方的職責����。在質量協(xié)議中明確雙方的溝通方式、溝通頻次以及相關要求����,以便及時解決問題、做到有據(jù)可查。在建設MAH文件體系時����,也要注意把握好一定的“度”。如果過于嚴苛����,可能會影響項目的運轉效率和進度;如果過于寬松����,可能無法確保項目的質量。因此����,我們需要在“度”的掌握上找到一個平衡點。
綜上所述����,建設MAH文件體系的關鍵在于“合規(guī)”和“合理”����,在遵守藥品管理法規(guī)前提下,MAH通過對受托方的審計����,過程中加強溝通與合作����,確保雙方能夠及時交流����、明確責任,根據(jù)自身實際情況在保障雙方權益和不影響實際執(zhí)行的基礎上����,建立雙方認可的文件管理體系。文件的實際執(zhí)行與文件的要求保持一致是非常重要的����,只有這樣,我們才能夠有效地推動項目的實施����,確保藥品的質量、安全和合規(guī)性����。
參考文獻
[1]《藥品生產(chǎn)監(jiān)督管理辦法》(2020年第28號).國家藥品監(jiān)督管理局,2020-01-22.
[2]《藥品上市許可持有人檢查要點》(征求意見稿).國家藥品監(jiān)督管理局����,2022-03-31.
[3]《中華人民共和國藥品管理法藥品管理法》.國家市場監(jiān)督管理總局����,2019-08-26.
[4] MAH | 國家局檢查要點擬發(fā)布,MAH如何創(chuàng)建和維護合規(guī)的質量保證體系文件����?[EB/OL].(2022-04.08).http://www.phirda.com/artilce_27297.html.