過程分析技術(shù)(process analytical technology�,PAT)是指以保證終產(chǎn)品質(zhì)量為目的�,通過對有關(guān)原料����、生產(chǎn)中物料及工藝的關(guān)鍵參數(shù)和性能指標(biāo)進(jìn)行實(shí)時(shí)(即在工藝過程中)檢測的一個(gè)集設(shè)計(jì)、分析和生產(chǎn)控制為一體的系統(tǒng)����。
它最早起源于 1993 年美國分析化學(xué)家協(xié)會(huì)(AOAC International)發(fā)起的一個(gè)論壇����,后在 2001 年由美國食品藥品監(jiān)督管理局 (Food and Drug Administration�,F(xiàn)DA)藥品評(píng)價(jià)與研究中心主任 Janet Woodcock 博士總結(jié)提出了 PAT 的倡 議。
2004 年 FDA 正式發(fā)表了關(guān)于 PAT 的工業(yè)指導(dǎo)原則——Guidance for industry PAT——A framework for innovative pharmaceutical development�,manufacturing, and quality assurance�,拉開了 PAT 在制藥領(lǐng)域應(yīng)用的序幕。
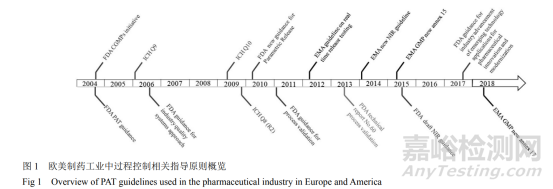
該技術(shù)不僅可以縮短工時(shí)����、減少誤差�,還可以使監(jiān)管更加有據(jù)可依,因此越來越多的歐美制藥企業(yè)開始引入 PAT 以實(shí)現(xiàn)藥品生產(chǎn)過程的全程監(jiān)控���,從而保證產(chǎn)品的質(zhì)量。同時(shí)����,為了推廣和規(guī)范 PAT 技術(shù),這些國家和地區(qū)的藥品監(jiān)管部門�,也先后出臺(tái)了許多相關(guān)的標(biāo)準(zhǔn)和指導(dǎo)原則(見圖 1)�。
本文將目前歐美等國家與 PAT 概念����、方法����、應(yīng)用和監(jiān)管相關(guān)的主要標(biāo)準(zhǔn)和指導(dǎo)原則加以總結(jié)和分析���,以幫助國內(nèi)制藥領(lǐng)域更好地理解和應(yīng)用這項(xiàng)技術(shù)。
美國的相關(guān)舉措
FDA 作為全球制藥領(lǐng)域的風(fēng)向標(biāo)�,成為最早發(fā)起在制藥領(lǐng)域?qū)嵤?PAT 的監(jiān)管機(jī)構(gòu)����。
FDA 在制藥領(lǐng)域提出 PAT 的倡議,并非心血來潮�,而是各方利益沖突的撞擊與發(fā)展����。
隨著制藥行業(yè)的迅速發(fā)展,美國制藥工業(yè)界認(rèn)為 FDA 過于嚴(yán)格���,使企業(yè)在生產(chǎn)過程中缺乏靈 活性����;而 FDA 則面臨人員短缺但藥物申請的文件審查�、 現(xiàn)場認(rèn)證以及突發(fā)事件處理等越來越繁重的負(fù)擔(dān)�,加之公眾對于高質(zhì)量藥品的期待���,為了協(xié)調(diào)三方的矛盾�, FDA 在 2002 年發(fā)起了一個(gè)新的倡議——Pharmceutical CGMPs for the 21st century——a risk-based approach����,并于 2004 年形成最終報(bào)告����,告知制藥企業(yè)和公眾,F(xiàn)DA 將 建立更加科學(xué)的基于風(fēng)險(xiǎn)管理的高效監(jiān)管新策略���,鼓勵(lì)制藥企業(yè)使用新的技術(shù):
如 PAT 來促進(jìn)藥品的連續(xù)生產(chǎn)以及質(zhì)量的提高和創(chuàng)新,并成立 PAT 工作組加強(qiáng)監(jiān)管 部門與制藥企業(yè)相關(guān)方面的溝通和合作����,推進(jìn) PAT 的 實(shí)施。該報(bào)告還首次提出了藥品生產(chǎn)領(lǐng)域一個(gè)全新的理 念——質(zhì)量源于設(shè)計(jì)(Quality by Design���,QbD)���,該理念為 PAT 等創(chuàng)新技術(shù)的實(shí)施提供了監(jiān)管和技術(shù)架構(gòu)����。
隨后 FDA 在 2004 年發(fā)表了 PAT 工業(yè)指導(dǎo)原則 Guidance for industry PAT——A framework for innovative pharmaceutical development,manufacturing�, and quality assurance。
該指導(dǎo)原則首先介紹了在制藥領(lǐng)域推行 PAT 的背景����、目的以及 PAT 的定義�,然后系統(tǒng)地介紹了制藥企業(yè)實(shí)施 PAT 需要獲得哪些方面的知識(shí),F(xiàn)DA 為了推行 PAT 做了哪些努力(包括成立 PAT 專家組����,PAT 培訓(xùn)等等)�,以及制藥企業(yè)應(yīng)如何與 FDA 合作、溝通����、提交資料以獲得實(shí)施 PAT 的批準(zhǔn)。
從該指導(dǎo)原則可知�,PAT 不是一項(xiàng)單純的技術(shù)而是一 個(gè)技術(shù)領(lǐng)域����,制藥企業(yè)實(shí)施 PAT 不僅需要對生產(chǎn)過程有深刻的理解���,還需要了解和掌握與 PAT 相關(guān)的知識(shí) 和工具。
PAT 工具主要包括以下四類:用于設(shè)計(jì)����、數(shù)據(jù)采集和過程分析的多變量分析工具;現(xiàn)代過程分析儀器和過程分析化學(xué)工具����;過程控制工具以及持續(xù)的改進(jìn)和知識(shí)管理工具。另外在實(shí)施 PAT 時(shí)還應(yīng)掌握風(fēng)險(xiǎn)管理方法���、系統(tǒng)集成方法和實(shí)時(shí)放行方法�?���! ?/span>
其實(shí)早在 FDA 提出 PAT 實(shí)時(shí)放行方法以前,在歐美等無菌藥品生產(chǎn)領(lǐng)域已經(jīng)用實(shí)際行動(dòng)在踐行這一理念——參數(shù)放行(Parametric release)�。
所謂參數(shù)放行就是一種基于對業(yè)已通過驗(yàn)證的滅菌程序的有效質(zhì)量控制、監(jiān)測和系統(tǒng)文件管理來替代依賴于最終產(chǎn)品檢驗(yàn)的無菌放行程序���。
由于無菌檢驗(yàn)方法的統(tǒng)計(jì)學(xué)限制���,最終產(chǎn)品的無菌檢驗(yàn)只能提供一個(gè)發(fā)現(xiàn)無菌保證系統(tǒng)錯(cuò)誤的機(jī)會(huì)���,無菌藥品的無菌特性并非依賴于最終成品的檢驗(yàn),而是取決于對藥品生產(chǎn)全過程嚴(yán)格的質(zhì)量管理���。
1987 年����,F(xiàn)DA 正式頒布了參數(shù)放行的工業(yè)指南�,參數(shù)放行正式進(jìn)入藥品生產(chǎn)企業(yè)的生產(chǎn)質(zhì)量管理規(guī)范(Good Manufacturing Practices,GMP)�。
最新版本為 2010 年 2 月頒布的終端滅菌產(chǎn)品實(shí)施參數(shù)放行的相關(guān)申報(bào)資料要求 Guidance for industry submission of documentation in applications for parametric release of human and veterinary drug products terminally sterilized by moist heat processes,經(jīng)過多年的不斷完善����,發(fā)達(dá)國家的藥品監(jiān)管部門對于無菌藥品已經(jīng)普遍接受了參數(shù)放行的理念并付諸實(shí)踐�,分別制定了相應(yīng)的指導(dǎo)原則。
美國�、加拿大、澳大利亞等國家以及歐盟對參數(shù)放行的批準(zhǔn)及日常監(jiān)管內(nèi)容和形式基本一致���,都是在現(xiàn)行 GMP 管理的基礎(chǔ)上頒布參數(shù)放行指南和申報(bào)辦法�,由企業(yè)自愿申請�,藥品監(jiān)管部門進(jìn)行嚴(yán)格的資料審核和現(xiàn)場檢查后���,決定是否批準(zhǔn)企業(yè)的申請�,已獲批準(zhǔn)的藥品如發(fā)生重要因素的變更需重新申請?���! ?/span>
為了推行更加科學(xué)的監(jiān)管理念����、鼓勵(lì)制藥企業(yè)不斷創(chuàng)新和發(fā)展,F(xiàn)DA 也一直在積極地尋求與國內(nèi)外研究���、生產(chǎn)和監(jiān)管領(lǐng)域的合作���。
當(dāng) FDA 提出 QbD 理念后,很快被人用藥品注冊技術(shù)規(guī)定國際協(xié)調(diào)會(huì)(The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH) 所 采 納���, 與 FDA 聯(lián)合發(fā)布了 ICH Q8(R2)(Guidance for industry Q8(R2)Pharmaceutical development)、ICH Q9(Guidance for industry Q9 Quality risk management) 和 ICH Q10(Pharmaceutical quality system)�,這 3 個(gè)指導(dǎo)原則具體表達(dá)了過程和產(chǎn)品開發(fā)、 技術(shù)轉(zhuǎn)移和商品生產(chǎn)中 QbD 的理念���。
所謂 QbD 的理念就是從產(chǎn)品和生產(chǎn)工藝的研發(fā)階段開始進(jìn)行合理的實(shí)驗(yàn)設(shè)計(jì)���,研究產(chǎn)品質(zhì)量屬性與原料質(zhì)量屬性和生產(chǎn)過程參數(shù)之間的關(guān)系,建立能滿足產(chǎn)品性能且工藝穩(wěn)定的設(shè)計(jì)空間�,從而在生產(chǎn)階段建立一種可以在一定范圍內(nèi)調(diào)節(jié)偏差來保證產(chǎn)品質(zhì)量穩(wěn)定性的措施,并在商業(yè)化大生產(chǎn)開始后對工藝進(jìn)行連續(xù)的改進(jìn)����。
由此可見,盡管 QbD 不等同于 PAT���,但 PAT 是實(shí)現(xiàn) QbD 優(yōu)點(diǎn)的實(shí)質(zhì)要素和關(guān)鍵驅(qū)動(dòng)因素�。其中 ICH Q8(R2)系統(tǒng)地介紹了 QbD 的基本原則以及在藥品及其生產(chǎn)工藝的研發(fā)過程中如何應(yīng)用科學(xué)策略和質(zhì)量風(fēng)險(xiǎn)管理策略獲取對產(chǎn)品和生產(chǎn)工藝的全面理解���,從而細(xì)化了 PAT 實(shí)施前需要對生產(chǎn)過程深刻理解的具體范圍和要求���。
同時(shí) ICH Q8(R2)在 PAT“實(shí)時(shí)放行”方法后加上“檢測”二字����, 即實(shí)時(shí)放行檢測�,將關(guān)注點(diǎn)轉(zhuǎn)移到測量方法上,相比成品檢測�,實(shí)時(shí)放行檢測在生產(chǎn)過程中進(jìn)行質(zhì)量監(jiān)測, 將質(zhì)量控制從生產(chǎn)過程的終點(diǎn)向上游轉(zhuǎn)移�,明確了 PAT 的優(yōu)勢。
ICH Q9 重點(diǎn)介紹了質(zhì)量風(fēng)險(xiǎn)管理的原則和常用分析工具����,它是實(shí)施 PAT 所需的理論基礎(chǔ)之一�, 但其內(nèi)容適用范圍不只局限于 PAT。
盡管 PAT 等新興的技術(shù)前景可觀���,但制藥企業(yè)仍會(huì)對采用新技術(shù)猶豫不決����,主要原因是擔(dān)心具體監(jiān)管措施不明朗而導(dǎo)致新技術(shù)的應(yīng)用延遲�。為此,F(xiàn)DA 也在 PAT 實(shí)施細(xì)則方面進(jìn)行了很多努力����。
2006 年����,F(xiàn)DA 發(fā)布了 制藥企業(yè) CGMP 規(guī)范的質(zhì)量體系工業(yè)指南——Quality systems approach to pharmaceutical CGMP regulations�, 旨在幫助制藥企業(yè)建立以科學(xué)和風(fēng)險(xiǎn)管理為基礎(chǔ)的質(zhì)量體系來滿足現(xiàn)行動(dòng)態(tài)生產(chǎn)質(zhì)量管理規(guī)范(Current Good Manufacturing Practices���,CGMP)的要求。
該指南建議制藥企業(yè)建立包含管理職責(zé)����、資源、生產(chǎn)運(yùn)作和評(píng)估活動(dòng) 4 個(gè)要素在內(nèi)的全面質(zhì)量體系�,并對每個(gè)要素的組成�、職責(zé)進(jìn)行了詳細(xì)的建議。
FDA 認(rèn)為擁有完善質(zhì)量體系和恰當(dāng)生產(chǎn)工藝的制藥企業(yè)能夠進(jìn)行多種類型的改進(jìn)�,能夠?yàn)?ldquo;設(shè)計(jì)的質(zhì)量”����、持續(xù)的改進(jìn)以及藥品生產(chǎn)過程中的風(fēng)險(xiǎn)管理提供必要的框架���。
在生產(chǎn)運(yùn)作要素內(nèi)���,F(xiàn)DA 認(rèn)為盡管對于某些生產(chǎn)過程可以用時(shí)間來界定終點(diǎn),但企業(yè)應(yīng)當(dāng)有能力利用在線參數(shù)建立生產(chǎn)控制����,這些參數(shù)是以實(shí)時(shí)檢測和監(jiān)測設(shè)備測定的預(yù)期工藝終點(diǎn)為基礎(chǔ)的����。
該指導(dǎo)原則為 PAT 在現(xiàn)行生產(chǎn)控制中的應(yīng)用找到了切入點(diǎn)并明確了其在質(zhì)量體系中所處的位置。
2011 年 FDA 整合了新的監(jiān)管理念�,發(fā)布了改版的工藝驗(yàn)證指南——Guidance for industry process validation:general principles and practices,工藝驗(yàn)證是法律規(guī)定制藥企業(yè)必不可少的生產(chǎn)活動(dòng)之一���,該指南對工藝驗(yàn)證的概念和范疇進(jìn)行了廣度和深度的拓展����,將 FDA 對于工藝驗(yàn)證的要求,從原先對 3 個(gè)生產(chǎn)規(guī)模批次樣品生產(chǎn)過程的驗(yàn)證�,變成了涉及整個(gè)產(chǎn)品生命周期和生產(chǎn)中發(fā)生的一系列活動(dòng)的驗(yàn)證。
其中 FDA 建議可以使用 PAT 的工藝控制策略�,由于 PAT 工藝被設(shè)計(jì)用來實(shí)時(shí)檢測一種在加工材料的多種屬性�,因此需要使用不同的工藝性能確認(rèn)方法����,重點(diǎn)關(guān)注待檢測屬性的檢測系統(tǒng)和控制環(huán)。
工藝性能確認(rèn)實(shí)際上就是為證明工藝可重現(xiàn)且可以始終如一的生產(chǎn)出優(yōu)質(zhì)產(chǎn)品尋找科學(xué)依據(jù)的過程���,但該指導(dǎo)原則對于 FDA 對工藝驗(yàn)證不同階段文件的要求和期望并未給出具體的描述。
2013 年美國注射劑協(xié)會(huì)(Parenteral Drug Association���,PDA) 發(fā)布了題為 Technical report No.60 process validation:A lifecycle approach 的第 60 號(hào)技術(shù)報(bào)告,對 FDA 關(guān)于產(chǎn)品生命周期內(nèi)工藝驗(yàn)證的全過程進(jìn)行了具體的詮釋����。
該技術(shù)報(bào)告的核心理念是“工藝驗(yàn)證不是一個(gè)一次性的活動(dòng)����,而是一個(gè)貫穿于整個(gè)產(chǎn)品生命周期,連接工藝開發(fā)與商業(yè)化生產(chǎn)�,并在日常商業(yè)化生產(chǎn)中維持的活動(dòng)”����。它為 FDA 2011 年版工藝驗(yàn)證指南在制藥領(lǐng)域?qū)嵤┊嫵隽司唧w的線路圖�。
該技術(shù)報(bào)告在第六部分——實(shí)施工藝驗(yàn)證的系統(tǒng)和技術(shù)專門用了一節(jié)講述 PAT 系統(tǒng)的選擇以及 PAT 系統(tǒng)設(shè)計(jì)階段工藝驗(yàn)證的考慮要點(diǎn)等制藥企業(yè)關(guān)注的具體問題����。
2015 年���,F(xiàn)DA 又發(fā)布了工業(yè)界開發(fā)和申報(bào)近紅外分析方法指導(dǎo)原則草案——Development and submission of near infrared analytical procedures,Guidance for industry����,Draft guidance——近紅外分析方法是 PAT 最常用的分析技術(shù)之一����,該指導(dǎo)原則給出了制藥行業(yè)在新藥申請、簡化新藥申請以及藥物主文件中采用近紅外分析方法的開發(fā)�、驗(yàn)證和變更需要考慮的問題以及需提交的信息���。
同年,F(xiàn)DA 藥品審評(píng)與研究中心下屬的藥品質(zhì)量辦公室還啟動(dòng)了新興技術(shù)項(xiàng)目(Emerging Technology Program)���,期望制藥企業(yè)從早期就能參與 該項(xiàng)目���,通過與 FDA 的新興技術(shù)團(tuán)隊(duì)召開更多會(huì)議、 進(jìn)行更多溝通探討���,共同研究新生產(chǎn)技術(shù)中的相關(guān)問題,從而在提交注冊資料之前就能發(fā)現(xiàn)并解決這些問題�。
FDA 更是于 2017 年發(fā)布了新興技術(shù)應(yīng)用的先進(jìn)性使得藥品生產(chǎn)基礎(chǔ)現(xiàn)代化的工業(yè)指南——Guidance for industry advancement of emerging technology applications for pharmaceutical innovation and modernization�,對制藥企業(yè)如何參與 FDA 的新興技術(shù)項(xiàng)目來推動(dòng)包括 PAT 等新興生產(chǎn)技術(shù)的應(yīng)用進(jìn)行了詳細(xì)解讀?��! ?nbsp;
美國藥典(The United States Pharmacopeia����,USP) 委員會(huì)是為在美國境內(nèi)生產(chǎn)和銷售的處方及非處方藥物�、食物補(bǔ)充劑和其他保健產(chǎn)品制訂質(zhì)量標(biāo)準(zhǔn)的法定機(jī)構(gòu),為協(xié)助 FDA 推進(jìn) PAT 技術(shù)����,美國藥典委員會(huì)也進(jìn)行了大量的工作。
其中兩項(xiàng)主要的工作是修訂現(xiàn)有 USP 標(biāo)準(zhǔn)中有關(guān)近紅外的章節(jié)(< 1119 > Near-Infrared Spectroscopy)���,強(qiáng)調(diào)了其作為 PAT 應(yīng)用的要求����;增 加了 PAT 另一項(xiàng)常用技術(shù)拉曼光譜的章節(jié)(< 1120 > Raman Spectroscopy)。
與近紅外光譜相似���,拉曼光譜具有快速、樣品預(yù)處理簡單等優(yōu)勢���,同時(shí)它受水分干擾 小,無需復(fù)雜建模使其逐漸成為 PAT 的新寵�。最新 版 USP41 通則 1120 對拉曼光譜的應(yīng)用范圍、影響因素�、儀器以及性能確認(rèn)等進(jìn)行了詳細(xì)的說明和要求�。
歐盟的相關(guān)舉措
歐盟作為制藥領(lǐng)域的又一發(fā)達(dá)地區(qū)�,其制定的標(biāo)準(zhǔn)和指導(dǎo)原則也經(jīng)常成為大家觀摩的范本。
2014 年歐洲藥品管理局(European Medicines Agency�,EMA) 發(fā)布正式版 Guideline on process validation for finished products information and data to be provided in regulatory submissions����,該指南在 2016 年 又 做 了 小 的 修 改����。
與 FDA 的工藝驗(yàn)證指南將工藝驗(yàn)證劃分為 3 個(gè)階段(工藝設(shè)計(jì)���、工藝確認(rèn)和持續(xù)工藝驗(yàn)證)不同���,EMA 的這項(xiàng)驗(yàn)證指南僅適用于制劑����,且僅對應(yīng)于 FDA 定義的第二階段——工藝確認(rèn)�。
在 EMA 的工藝驗(yàn)證指南中引入了連續(xù)工藝確認(rèn)(Continuous process verification���,CPV)�, 這一概念來源于 ICH Q8����,當(dāng)一種產(chǎn)品是通過 QbD 方法 開發(fā)并已經(jīng)科學(xué)地建立了能高度保證產(chǎn)品質(zhì)量的工藝控制時(shí),可以采用 CPV 替代傳統(tǒng)工藝驗(yàn)證�。
CPV 是通過建立一個(gè)工藝確證體系�,對物料屬性、關(guān)鍵質(zhì)量屬性和關(guān)鍵工藝參數(shù)建立了科學(xué)的控制策略,從而保證終產(chǎn)品的質(zhì)量���。
指南中明確指出:控制策略應(yīng)是經(jīng)過常規(guī)評(píng)估的,可以選用 PAT 和多變量統(tǒng)計(jì)過程控制(Multivariate statisitical process control�,MSPC)作為控制策略的工具。
對于不同的生產(chǎn)步驟還可以選擇采用傳統(tǒng)工藝驗(yàn)證和連續(xù)工藝驗(yàn)證混合的方式���,在申報(bào)文件中應(yīng)提供使用混合方法的論證并指出在哪些生產(chǎn)步驟采用了混合方法。
隨后的 2015 年 3 月歐盟委員會(huì)發(fā)布了最新改版的歐盟 GMP 附錄 15 確認(rèn)與驗(yàn)證 Volume 4 EU Guidelines for good manufacturing practice for medicinal products for human and veterinary use Annex 15:qualification and validation�,將工藝驗(yàn)證拓展到產(chǎn)品的全生命周期,對企業(yè)如何進(jìn)行確認(rèn)與驗(yàn)證進(jìn)行了具體的表述�,其中對于企業(yè)采用傳統(tǒng)工藝驗(yàn)證方法、連續(xù)工藝確認(rèn)方法或混合方法的表述與 EMA 制劑工藝驗(yàn)證指南基本一致���。
歐盟也是較早對無菌產(chǎn)品使用參數(shù)放行的地區(qū)之一����, 2012 年 EMA 頒布了新版的實(shí)時(shí)放行檢測指南 Guideline on real time release testing(formerly guideline on parametric release)以取代 2001 年頒布的參數(shù)放行指南。
該指南指出實(shí)時(shí)放行不再只適用于無菌藥物的滅菌過程���,而是適用于化學(xué)藥和生物制劑原料�、中間體和制劑的各個(gè)生產(chǎn)過程�。該指南還給出了實(shí)施實(shí)時(shí)放行的法規(guī)基礎(chǔ)、應(yīng)用 實(shí)例以及申請批準(zhǔn)需要的文件材料�。
2018 年 4 月歐盟委員會(huì)發(fā)布了最新改版的歐盟 GMP 附錄 17 實(shí)時(shí)放行檢測和參數(shù)放行 Volume 4 EU Guidelines for good manufacturing practice for medicinal products for human and veterinary use annex 17:real time release testing and parametric release�,同 樣在原有參數(shù)放行的基礎(chǔ)上增加了實(shí)時(shí)放行的內(nèi)容����,并對企業(yè)如何實(shí)施實(shí)時(shí)放行給出了具體的要求?���! ?/span>
關(guān)于近紅外光譜技術(shù)在制藥企業(yè)應(yīng)用的探討在歐洲起步較早,英國�、荷蘭�、法國等研究機(jī)構(gòu)都有過相關(guān)的應(yīng)用指導(dǎo)原則出臺(tái),其中 EMA 制定的制藥工業(yè)近紅外光譜技術(shù)應(yīng)用�、申報(bào)和變更資料要求指南 Guideline on the use of near infrared spectroscopy by the pharmaceutical industry and the data requirements for new submissions and variations 在 2003 年發(fā)布后又根據(jù)法規(guī)與技術(shù)的變化進(jìn)行了更新,于 2014 年發(fā)布新版���。
它在建立、驗(yàn)證����、變更近紅外方法方面為藥品生產(chǎn)企業(yè)提供了詳細(xì)的指導(dǎo)����,并規(guī)定了在新藥申報(bào)或方法變更時(shí)����,使用近紅外方法需要提交的數(shù)據(jù),該指導(dǎo)原則在內(nèi)容設(shè)置上較 FDA 的近紅外指導(dǎo)原則更加具體�,此次的更新版本還特別在方法通用要求���、定性 / 定量模型分類要求等方面針對 PAT 的應(yīng)用進(jìn)行了說明����?! ?nbsp;
歐洲藥典(The European Pharmacopoeia,EP) 是歐洲藥品質(zhì)量檢測的唯一指導(dǎo)文獻(xiàn)�,它由歐洲藥品質(zhì)量管理局(European Directorate for the Quality Control of Medicines���,EDQM)出版發(fā)行���。EP 是最早收錄近紅外光譜技術(shù)(2.2.40. Near-infrared spectroscopy)的藥典���。
2018 年 1 月���,EDQM 又在歐洲藥典論壇上發(fā)布了 PAT 草案(5.25. Process analytical technology)���,計(jì)劃將其收入 EP����。該草案對 PAT 的定義����、測量方式以及數(shù)據(jù)的分析方法等進(jìn)行了簡單介紹。為了推動(dòng)和支持 PAT 技 術(shù)的應(yīng)用����,EDQM 還新增或修訂了 EP 中 PAT 相關(guān)的 9 個(gè)章節(jié)(見表 1),主要涉及 PAT 的常用技術(shù)與數(shù)據(jù)分析方法�。
草案也分別就這 9 個(gè)章節(jié)的修訂內(nèi)容進(jìn)行了介紹。
如1. 通則的修訂:1.1 部分說明一個(gè)物料可基于產(chǎn)品設(shè)計(jì)結(jié)合其控制策略和從生產(chǎn)工藝驗(yàn)證研究中所生成的數(shù)據(jù)證明其具備藥典質(zhì)量����。同時(shí)還說明加強(qiáng)質(zhì)量控制的方法可利用 PAT 和 / 或?qū)崟r(shí)放行檢測策略(包括參數(shù)放行)替代僅檢測終產(chǎn)品的方法���。
再如章節(jié) 2.2.40 近紅外光譜部分:增加近紅外近線、在線和原位測試的表述����, 同時(shí)在樣品準(zhǔn)備 / 代表性部分增加了清除樣品以及在不同測量模式中使用光纖探頭系統(tǒng)。如果轉(zhuǎn)移光纖探頭用于背景光譜測量困難����,可以使用內(nèi)置參照用于過程分析,同時(shí)還根據(jù)儀器的測量模式和使用位點(diǎn)提供了詳細(xì)的儀器性能控制方法����。另外在定性分析部分除常規(guī)的鑒別分析外,還介紹了限度分析(如用于控制干燥終點(diǎn)) 和趨勢分析(如混合均勻性監(jiān)測)����。
總結(jié)與展望
由上述歐美制藥領(lǐng)域 PAT 相關(guān)的標(biāo)準(zhǔn)和指導(dǎo)原則可見,PAT 技術(shù)自 2002 年 FDA 在制藥領(lǐng)域提出至今���,已經(jīng)逐步從最初的設(shè)想向?qū)嵱没较蜻~進(jìn)����。
我國在該領(lǐng)域的應(yīng)用起步較晚�,且大都集中在中藥的過程控制領(lǐng)域���。盡管 2016 年發(fā)布的《醫(yī)藥工業(yè)發(fā)展規(guī)劃指南》和 《智能制造工程實(shí)施指南(2016 - 2020)》均將 PAT 作為未來醫(yī)藥領(lǐng)域發(fā)展主要任務(wù)之一,但有關(guān) PAT 的具體應(yīng)用指南和監(jiān)管措施鮮有發(fā)布���。
現(xiàn)行《中國藥典》(2015 年版)雖已經(jīng)收錄近紅外光譜和拉曼光譜技術(shù)���,但在內(nèi)容設(shè)置上主要還是針對成品的定性和定量分析,未涉及藥品生產(chǎn)過程的監(jiān)控環(huán)節(jié)����。
由于多種因素的制約���,無論是 PAT 技術(shù)在制藥領(lǐng)域的應(yīng)用還是監(jiān)管�,我國與歐美之間仍然存在很大的差距���。相信���,隨著 QbD 理念的不斷深入,制藥企業(yè)終將理解以風(fēng)險(xiǎn)管理為基礎(chǔ)的 PAT 技術(shù)是了解生產(chǎn)工藝����、控制產(chǎn)品關(guān)鍵質(zhì)量屬性的最有效手段���。
可以預(yù)見,隨著國內(nèi)制藥領(lǐng)域 PAT 的深入開展���, 藥品生產(chǎn)企業(yè)和監(jiān)管部門對 PAT 技術(shù)的逐步采納和認(rèn)可�,其在藥物分析領(lǐng)域的標(biāo)準(zhǔn)和原則也將不斷完善和成熟����。
來源:《中南藥學(xué)》2019 年 9 月 第 17 卷 第 9 期
原標(biāo)題:《歐美制藥工業(yè)中過程控制主要標(biāo)準(zhǔn)和指導(dǎo)原則簡介》
作者:馮艷春,肖亭���,胡昌勤(中國食品藥品檢定研究院)