質量管理模塊作為GMP檢查六大體系(質量��、實驗室、生產(chǎn)、設施及設備、物料系統(tǒng)�、包裝和標簽)的重要環(huán)節(jié)及核心內容��,是支撐的藥品質量體系的基石����。ICH Q10中描述藥物質量體系主要包括四個要素��,分別是工藝性能和產(chǎn)品質量監(jiān)督體系�、糾正和預防措施體系(CAPA)、管理變更體系��、工藝性能和產(chǎn)品質量的管理審核體系,CAPA是藥品質量體系的重要組成部分�。
CAPA于2006年首次由FDA正式引入制藥行業(yè)。CAPA是指對存在的或潛在的不合格或不期望情況的原因進行調查分析����,采取措施以防止問題再發(fā)生或避免發(fā)生的全部活動。CAPA不僅是就事論事的對單個問題(偏差�、OOS)情況的處理,更是從根本上調查產(chǎn)生問題的原因����,采取適當和有效的糾正和/或預防措施以防止其再次發(fā)生。對于藥企來說����,涉及影響藥品質量和安全的各方面活動都可能觸發(fā)CAPA程序。沒有CAPA��,質量體系就無法正常����、高效地運行,藥企實施CAPA的目的在于對質量體系不斷地完善和改進�,從而提高企業(yè)質量管理水平,是企業(yè)長期發(fā)展的必然要求����。
一��、CAPA概念與法規(guī)
根據(jù)FDA的描述��,CAPA的目的是收集信息����、分析信息�、識別和調查產(chǎn)品和質量問題,并采取適當和有效的糾正和/或預防措施以防止其再次發(fā)生�。驗證CAPA,與負責人溝通CAPA活動��、為管理評審提供相關信息�,以及記錄這些活動,對于有效處理產(chǎn)品和質量問題����、防止其再次發(fā)生以及預防或最大限度減少各類故障至關重要。
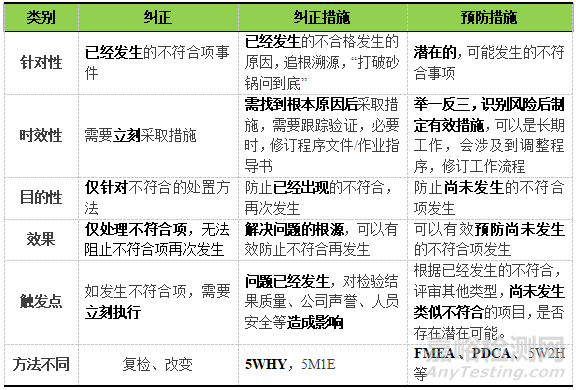
CAPA通常分為三種類型����,分別是糾正�、糾正措施�、預防措施����,定義分別如下(參考ISO 9000:2005(E)):
●糾正(Correction):“action to eliminate a detected nonconformity”.(為消除已發(fā)現(xiàn)的不符合項所采取的措施);
●糾正措施(Corrective action):“action to eliminate the cause of a detected nonconformity or other undesirable situation.”(為消除已發(fā)現(xiàn)的不合格或其他不期望情況的原因所采取的措施)����;
●預防措施(preventive action):“action to eliminate the cause of a potential nonconformity or other undesirable situation.”(為消除潛在不合格或其他潛在不期望情況的原因所采取的措施)。
在實際管理過程中需要厘清概念的差異并嚴格區(qū)分����,針對上述三個概念,可從時效性�、目的、效果��、觸發(fā)點等方面加以對比區(qū)分��,見表1��。
▲表1-CAPA三種類型的對比
1.2 CAPA相關法規(guī)
國內CAPA沒有專門的法規(guī)和指南����,但在與制藥相關的眾多與質量相關的法規(guī)指南中,或多或少均涉及CAPA體系的相關內容。以下是歸納了部分涉及與CAPA相關的法規(guī)����、指南或標準。
ISO 9001:2015質量管理體系標準:這是一個國際標準�,規(guī)定了組織應建立和維護的質量管理體系要求。其中包括了CAPA的要求�,包括問題識別、根本原因分析����、糾正措施和預防措施的實施、監(jiān)督和驗證等�。
FDA 21 CFR Part 820 – 質量管理體系法規(guī)QMSR:這個法規(guī)適用于美國制造和分發(fā)醫(yī)療器械的企業(yè),要求建立和維護適當?shù)馁|量體系��,包括CAPA流程��。
FDA 21 CFR Part 211 – 制劑藥品的cGMP:這個法規(guī)是美國版的GMP�,適用于美國藥品制造業(yè),規(guī)定了質量管理體系的要求�,包括CAPA的實施要求。
ICH Q10 制藥質量體系:第3節(jié) 工藝性能與持續(xù)改進(3.2 制藥質量體系要素����;3.2.2 糾正措施與預防措施體系)中描述“對于由調查研究投訴��,產(chǎn)品否決,不合格����,召回,偏差�,審計,官方檢查和發(fā)現(xiàn)����,及工藝性能趨勢和產(chǎn)品質量監(jiān)控等而產(chǎn)生的糾正措施和預防措施,制藥企業(yè)應有體系來執(zhí)行這些措施��。”并進一步提出調查問題的要求和CAPA實施目標�。
我國《藥品生產(chǎn)質量管理規(guī)范》(2011版):第十章 質量控制與質量保證(第六節(jié) 糾正措施和預防措施;第二百五十二條至第五十三條)�,分別敘述了中國藥監(jiān)對制藥企業(yè)建立CAPA系統(tǒng)的總體要求和實施內容。
二��、CAPA來源��、管理范圍及其應用
CAPA通過改進和優(yōu)化質量管理體系��,采取風險防控措施來防范潛在的安全風險�。通過CAPA的全面應用�,藥品制造企業(yè)可以保持質量和安全的持續(xù)改進����,更好地滿足監(jiān)管要求,并為患者提供安全有效的藥品����。
2.1 CAPA來源
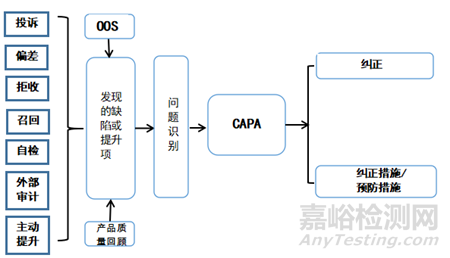
引發(fā)CAPA的問題可能源自質量管理體系內的各個方面。包括供應鏈管理�、制造過程、設備維護����、人力資源和培訓等,羅列制藥企業(yè)CAPA的通常來源�,見圖1.
▲圖1-制藥企業(yè)CAPA通常來源
無論出于何種原因或在何處啟動CAPA,都應始終從確定問題的那一刻開始立即采取糾正行動����,以穩(wěn)定局勢并防止其進一步影響生產(chǎn)和產(chǎn)品質量。
2.2 CAPA管理范圍及應用
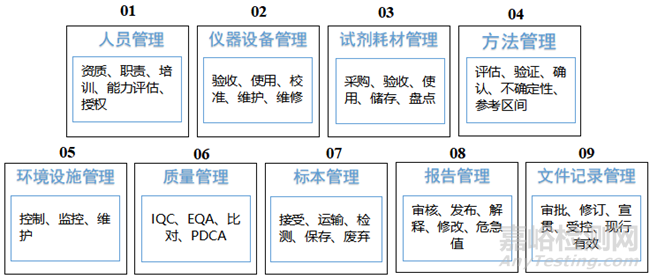
CAPA程序用于解決已經(jīng)發(fā)生的偏差或OOS�,并采取措施避免將來出現(xiàn)同樣的問題。CAPA的管理范圍覆蓋了GMP的各個環(huán)節(jié)��,包括人員培訓�、儀器設備管理��、耗材試劑管理��、方法管理等�,詳見圖2�。
▲圖2-CAPA管理范圍
ICH Q10描述藥品生命周期包括四個階段:藥品開發(fā)��、技術轉移�、商業(yè)化生產(chǎn)、產(chǎn)品終止��,CAPA在藥品全生命周期管理中扮演著關鍵的角色�。通過糾正和預防措施來管理風險、處理非合規(guī)事件�、持續(xù)改進質量,并確保符合法規(guī)要求����,從而確保藥品的安全性、有效性和合規(guī)性��。ICH Q10描述中進一步歸納了CAPA在藥品生命周期中的應用����,見表2��。
▲表2-CAPA系統(tǒng)在整個產(chǎn)品生命周期內的應用
三�、CAPA常規(guī)流程和實施
CAPA是制藥企業(yè)質量管理中常用的工具�,能夠很好地解決問題并預防再次發(fā)生。在實施CAPA時��,要確保流程規(guī)范化�、員工參與和培訓、數(shù)據(jù)收集和分析的有效性�,并持續(xù)監(jiān)控和改進。
3.1 CAPA常規(guī)流程
我國《藥品生產(chǎn)質量管理規(guī)范》(2011版)第六節(jié):糾正措施和預防措施�,第二百五十三條企業(yè)應建立實施糾正和預防措施的操作規(guī)程,內容包括:
1)對投訴����、召回、偏差����、自檢或外部檢查結果工藝性能和質量監(jiān)測趨勢以及其它來源的質量數(shù)據(jù)進行分析,確定已有和潛在的質量問題��。必要時��,應采用適當?shù)慕y(tǒng)計學方法����;
2)調查與產(chǎn)品����、工藝和質量保證系統(tǒng)有關的原因��;
3)確定所需采取的糾正和預防措施��,防止問題的再次發(fā)生����;
4)評估糾正和預防措施的合理性��、有效性和充分�;
5)對實施糾正和預防措施過程中所有發(fā)生的變更應予以記錄;
6)確保相關信息已傳遞到質量受權人和預防問題再次發(fā)生的直接負責人����;
7)確保相關信息及其糾正和預防措施已通過高層管理人員的評審。
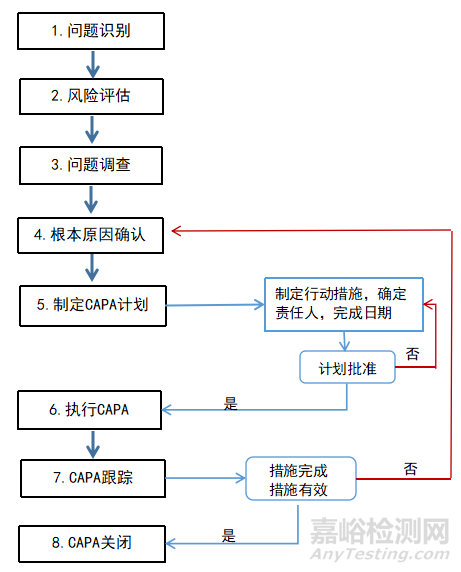
從上述文字可以看出我國GMP對藥企CAPA的執(zhí)行要求和內容做出了框架式的總體規(guī)定����,執(zhí)行層面需要企業(yè)在遵守法規(guī)指南的基礎上,根據(jù)實際情況具體實施����。參考ISO 9001��、FDA 21 CFR Part 820等相關標準和法規(guī)��,結合個人的經(jīng)驗和理解����,繪制藥企CAPA系統(tǒng)常規(guī)流程��,見圖3��。
▲圖3-CAPA常規(guī)實施流程
由上圖可看出��,制藥企業(yè)在實施CAPA時����,首先,要識別和評估問題�;其次,要通過問題調查����,確定其根本原因;再次,制定糾正措施來解決問題�,并實施預防措施以防止問題再次發(fā)生。在實施CAPA過程中��,還需要監(jiān)督和驗證措施的執(zhí)行����,并記錄所有的活動和結果,跟蹤并評價實施效果�,措施完成并且有效,則CAPA關閉�;否則,返回流程繼續(xù)查找根本原因�,依次執(zhí)行后續(xù)流程。
3.2 CAPA實施
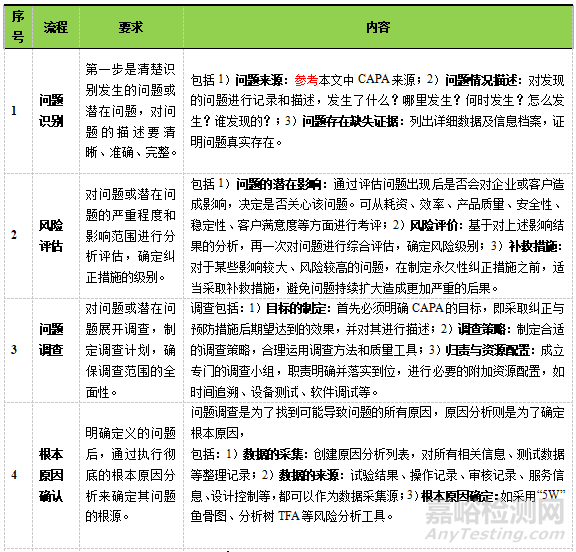
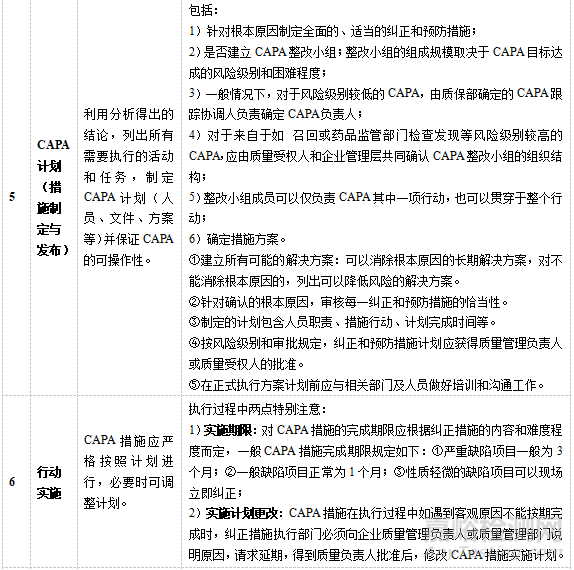
CAPA是質量管理體系的重要組成部分�,無論從政府監(jiān)管的需要還是企業(yè)自身產(chǎn)品質量控制以及客戶滿意度等方面來說�,CAPA的有效貫徹實施都是制藥企業(yè)質量管理體系中不容忽視的內容。結合CAPA常規(guī)流程��,進一步分析和闡述流程具體實施的要求和內容��,見表3�。
▲表3-CAPA實施要求與內容
四、總結
在GMP體系下��,CAPA實施流程和策略對于確保藥品的質量和合規(guī)性至關重要。CAPA流程包括問題識別��、問題調查��、糾正措施的實施��、預防措施的制定和驗證等步驟����。為了有效實施CAPA,必須制定一系列策略�,例如明確定義責任和角色、建立合適的糾正和預防方法��、建立持續(xù)改進和監(jiān)督機制等��。此外����,合適的培訓和溝通也是成功實施CAPA的關鍵因素。最終目標是持續(xù)改進和提高質量����,確保藥品的安全性和有效性。通過嚴格遵循CAPA實施流程和策略����,制藥企業(yè)能夠識別和解決問題��,防止其再次發(fā)生�,并符合相關法規(guī)和標準的要求����。只有致力于不斷改進CAPA流程和策略,并將其納入企業(yè)的質量體系中����,才能實現(xiàn)持續(xù)質量提升的目標。
參考文獻
[1]ICH Q10制藥質量體系(2008).
[2]《藥品生產(chǎn)質量管理規(guī)范》(2011版).
[3]ISO 9001:2015質量管理體系標準.
[4]FDA 21 CFR Part 211 –制劑藥品的cGMP(2023).
[5]FDA 21 CFR Part 820 – 質量管理體系法規(guī)QMSR(2023).
[6]丁曉玥,梁毅.制藥企業(yè)質量管理體系中糾正與預防措施(CAPA)的實施[J].機電信息��,2011(5):5.