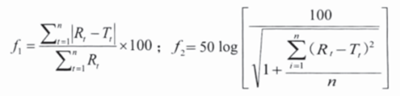溶出度試驗(yàn)技術(shù)是評(píng)價(jià)口服固體制劑內(nèi)在質(zhì)量的一種重要手段�,該試驗(yàn)不僅可為建立體內(nèi)外相關(guān)性提供基礎(chǔ)數(shù)據(jù)���,而且有望成為通過(guò)體外試驗(yàn)評(píng)價(jià)口服固體制劑質(zhì)量的簡(jiǎn)易�、有效可行的方法�。
在多種 pH 溶出介質(zhì)中溶出曲線的測(cè)定是先進(jìn)國(guó)家藥物審評(píng)機(jī)構(gòu)評(píng)價(jià)口服固體制劑內(nèi)在質(zhì)量的一種重要手段。
該試驗(yàn)可用于評(píng)估不同來(lái)源的同一制劑內(nèi)在質(zhì)量差異�。日本自 1998 年開始實(shí)施“藥品品質(zhì)再評(píng)價(jià)工程”,陸續(xù)出版了《醫(yī)療用醫(yī)薬品品質(zhì)情報(bào)集》( 即日本參比制劑目錄�、橙皮書�、Orange Book)�,其中詳細(xì)羅列了所收載制劑的四條標(biāo)準(zhǔn)溶出曲線。
美國(guó) FDA 藥品審評(píng)中心的仿制藥辦公室屬下的生物等效部也于2004年1月起���,在其官方網(wǎng)站上推出了“固體制劑溶出曲線數(shù)據(jù)庫(kù)”���。
目前我國(guó)雖未推出“溶出曲線數(shù)據(jù)庫(kù)”,但對(duì)口服固體制劑多條溶出曲線的測(cè)定已愈發(fā)受到關(guān)注與重視�。
本文在參照國(guó)際藥物聯(lián)合會(huì) (FIP) 頒布的《口服固體制劑溶出度試驗(yàn)指導(dǎo)原則》、日本厚生勞動(dòng)省頒布的《仿制藥生物等效性試驗(yàn)指導(dǎo)原則》�、FDA 頒布的《常規(guī)口服固體制劑溶出度試驗(yàn)工業(yè)指南》、美國(guó)藥典 31 版《溶出度試驗(yàn)驗(yàn)證法:研究與建立》( 附錄 1092) 和歐洲藥典 2008 年版《溶出度試驗(yàn)法》( 附錄溶出度試驗(yàn)法 2.9.3) 基礎(chǔ)上���,詳細(xì)闡述了如何采用多條溶出曲線評(píng)價(jià)固體制劑內(nèi)在質(zhì)量的具體試驗(yàn)步驟����,希望能通過(guò)此種檢測(cè)手段找到國(guó)產(chǎn)固體制劑與進(jìn)口原研制劑的差距����,為臨床療效的差異提供佐證,為國(guó)家相關(guān)技術(shù)法規(guī)的擬定與完善�、為企業(yè)切實(shí)有效地提高藥品質(zhì)量提供依據(jù)���。
一�、采用多條溶出曲線剖析參比制劑
原研制劑的確定
由于原研制劑 ( 亦稱參比制劑 ) 一般皆為國(guó)際制藥公司開發(fā)研制,在質(zhì)量水平及其控制上有嚴(yán)格的要求����。我國(guó)絕大多數(shù)藥物制劑均為仿制藥,只有質(zhì)量和原研制劑相同或相似����,才能保證其用藥的有效性和安全性。
其中溶出曲線的對(duì)比����,也應(yīng)以原研制劑作為“參比”。所以�,試驗(yàn)前一定要獲得原研制劑,以其作為參比制劑進(jìn)行測(cè)定���。
對(duì)于參比制劑的遴選����,原則上應(yīng)從多批號(hào)樣品中擷取一“標(biāo)準(zhǔn)批號(hào)”�,由于客觀條件的限制,建議最低要求應(yīng)獲取一個(gè)在有效期內(nèi)批號(hào)的樣品進(jìn)行研究����。
溶出介質(zhì)的選擇
1 普通制劑
酸性藥物制劑:pH 分別為 1.0 或 1.2����、5.5~6.5�、 6.8~7.5 和水。
中性或堿性藥物 / 包衣制劑:pH 分別為 1.0 或 1.2�、3.0~5.0、6.8 和水�。
難溶性藥物制劑:pH 分別為 1.0 或 1.2、4.0~4.5�、6.8 和水。
腸溶制劑:pH 分別為 1.0 或 1.2���、6.0����、6.8 和 水����。
以上根據(jù)藥物酸堿性而設(shè)定的相應(yīng) pH,參照了日本《仿制藥生物等效性試驗(yàn)指導(dǎo)原則》中的溶出度研究?jī)?nèi)容����。其中對(duì)難溶性藥物制劑則是根據(jù)美國(guó) FDA 公布的《仿制藥指導(dǎo)原則》�,但本文將 4.0 擴(kuò)展到了 4.5�。
2 緩 ( 控 ) 釋制劑
pH 分別為 1.0 或 1.2���、3.0~5.0���、6.8~7.5 和 水。
3 討論與說(shuō)明
藥物 pKa 小于 3.0 的可看作酸性藥物�,大于或等于 3.0 的可看作中 / 堿性藥物。
在進(jìn)行溶出曲線測(cè)定之前����,應(yīng)首先測(cè)定主成分在各種溶出介質(zhì)中的溶解度,以確定該制劑是否為 “pH 依賴性制劑”���。日本橙皮書中收載有這些數(shù)據(jù)�。
試驗(yàn)前還應(yīng)進(jìn)行原料藥在不同 pH 溶出介質(zhì)中的穩(wěn)定性考察�,以確保試驗(yàn)數(shù)據(jù)的準(zhǔn)確測(cè)定。日本橙皮書中收載有該部分內(nèi)容�。
以上含有 pH 范圍的,可分別按 0.5 或 1.0 間隔測(cè)試����。如溶出曲線差異較大���,應(yīng)考慮分別試驗(yàn);如無(wú)明顯差異���,酌情選擇即可���。
美國(guó)一般采用 1.0、4.5�、6.8 和水,日本一般采用 1.2�、4.0、6.8 和水����。無(wú)論何種制劑都不宜采用 pH 8.0 以上的介質(zhì)進(jìn)行試驗(yàn)。如確有必要�,應(yīng)提供充足理由。
FDA 公布的溶出度數(shù)據(jù)庫(kù)中����,只有阿維 A(acitretin) 膠囊采用 pH 9.6 溶出介質(zhì)。
但對(duì)于含有明膠交聯(lián)的制劑���,由于此類制劑的生物利用度與交聯(lián)度呈正相關(guān)�,在溶出介質(zhì)中加入適量酶,有助于評(píng)價(jià)該制劑的溶出度�。建議研究時(shí)可考慮添加酶以及酶的種類與濃度。
溶出介質(zhì)的配制
1 歐洲藥典
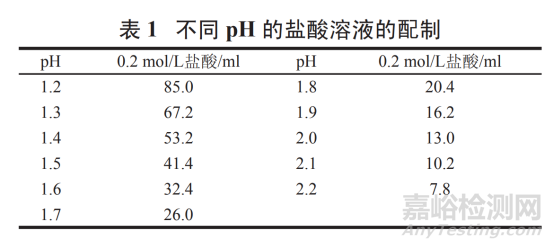
pH 1.0~2.2 鹽酸溶液:
精密量取濃鹽酸 9.0 ml���,加水稀釋至 1 000 ml,搖勻����,即得 pH 1.0 的鹽酸溶液。其他 pH 鹽酸溶液:按表 1 量取 0.2 mol/L 鹽酸 ( 精密量取濃鹽酸 18.0 ml�,加水稀釋至 1 000 ml,搖勻����,即得 ) 適量,加水稀釋至 200 ml���,搖勻���,即得 pH 1.2~2.2 鹽酸溶液。
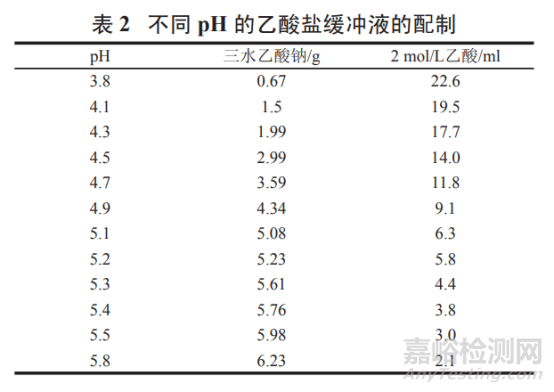
pH 3.8~5.8 乙酸鹽緩沖液:
按表 2 取三水乙酸鈉和 2 mol/L 乙酸 ( 取冰乙酸 120.0 g�,加水稀釋 至 1 000 ml,搖勻,即得 ) 適量���,加水溶解并稀釋至 1 000 ml����,搖勻�,即得。
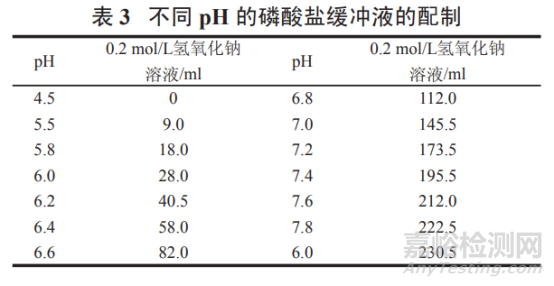
pH 4.5~8.0 磷酸鹽緩沖液:
精密稱取磷酸二氫鉀 6.80 g���,按表 3 加入 0.2 mol/L 氫氧化鈉溶液 ( 取 8.0 g 氫氧化鈉�,加水溶解并稀釋至 1000ml���,即得 ) 適量����,加水溶解并稀釋至 1 000 ml�,搖勻,即得����。
2 美國(guó)藥典
pH 1.0~2.2 鹽酸 / 氯化鉀溶液:
除需加入氯化鉀 0.75 g 外,其他配制方法同歐洲藥典�。
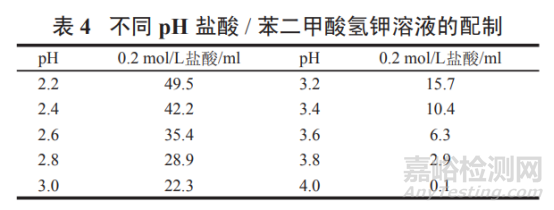
pH 2.2~4.0 鹽酸 / 苯二甲酸氫鉀溶液:
取苯二甲酸氫鉀 2.04 g�,按表 4 加入 0.2 mol/L 鹽酸適量���, 加水溶解并稀釋至 200 ml����,搖勻����,即得。
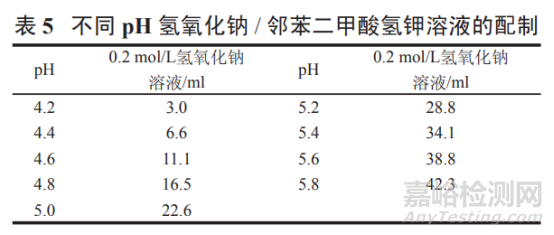
pH 4.2~5.8 氫氧化鈉 / 鄰苯二甲酸氫鉀溶液:
取鄰苯二甲酸氫鉀 2.04 g���,按表 5 加入 0.2 mol/L 氫氧化鈉溶液適量,加水溶解并稀釋至 200 ml���,搖勻���, 即得。
pH 5.8~8.0 氫氧化鈉 / 磷酸二氫鉀溶液:
同歐洲藥典�。
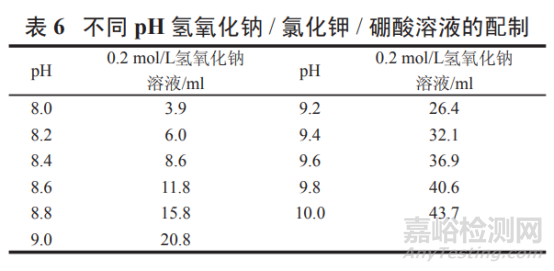
pH 8.0~10.0 氫氧化鈉 / 氯化鉀 / 硼酸溶液:
取氯化鉀 0.75 g 與硼酸 0.62 g,按表 6 加入 0.2 mol/L 氫氧化鈉溶液適量����,加水溶解并稀釋至 200 ml,搖勻,即得����。
3 日本藥典
pH 1.2 溶液:
取氯化鈉 2.0 g,加水適量溶解后�,加鹽酸 7 ml,再加水稀釋至 1 000 ml�,混勻, 即得�。
pH 4.0 溶液:
將 0.05 mol/L 乙酸溶液與 0.05 mol/L 乙酸鈉溶液按 16.4 ∶ 3.6 比例混合,即得���。
pH 3.0~6.0 介質(zhì) ( 除 4.0 外 ):
取十二水合磷酸氫二鈉 17.91 g�,加水溶解并稀釋至 1 000 ml���, 得 0.05 mol/L 磷酸氫二鈉溶液���。另取一水合枸櫞酸 5.25 g,加水溶解并稀釋至 1 000 ml����,得 0.025 mol/L 枸櫞酸溶液。用該枸櫞酸溶液調(diào)節(jié)上述磷酸氫二鈉溶液至所需 pH 即可���。
pH 6.8 磷酸鹽緩沖液:
取磷酸二氫鉀 1.7 g 和無(wú)水磷酸氫二鈉 1.775 g�,加水溶解并定容至 1 000 ml, 即得����。
4 中國(guó)藥典
配制方法詳見中國(guó)藥典 2005 年版附錄。
5 討論與說(shuō)明
建議根據(jù)原研制劑生產(chǎn)廠商的國(guó)別選取溶出介質(zhì)配制法���。各介質(zhì)配制 pH 誤差應(yīng)控制在 ±0.05���。
溶出曲線的測(cè)定
1 測(cè)定時(shí)間點(diǎn)和結(jié)束時(shí)間點(diǎn)的設(shè)定
對(duì)于測(cè)定時(shí)間點(diǎn),普通制劑與腸溶制劑可分別為 5�、10、15���、20、30���、45�、60����、90�、120 min�,此后每隔 1 h 測(cè)定。緩 / 控釋制劑可為 15�、30、45�、 60、90 min 和 2���、3���、4、5����、6、8���、10���、12、24 h����。
對(duì)于結(jié)束時(shí)間點(diǎn)����,在酸性介質(zhì) (pH 1.0~3.0) 中最長(zhǎng)測(cè)定時(shí)間為 2 h���;在其他各 pH 介質(zhì)中普通制劑與腸溶制劑均為 6 h���,緩 / 控釋制劑為 24 h。
但當(dāng)連續(xù)兩點(diǎn)溶出率均達(dá) 90%(緩 / 控釋制劑為 85% )����,且差值小于 5%時(shí),試驗(yàn)則可提前結(jié)束����。
2 討論與說(shuō)明
測(cè)定時(shí)間點(diǎn)的擬定:
研究表明,相當(dāng)一部分原研制劑的溶出曲線具有延遲���、拐點(diǎn)等現(xiàn)象 ( 如辛伐他汀片原研制劑就有5~10 min的延遲釋放 )。
提示:
在仿制研發(fā)與質(zhì)量評(píng)價(jià)時(shí)應(yīng)加以注意����,故以上測(cè)定時(shí)間點(diǎn)擬定得較為“緊湊”。但當(dāng)比較時(shí)間點(diǎn)確定后����,對(duì)于仿制制劑���,則可僅進(jìn)行比較時(shí)間點(diǎn)的測(cè)定。
試驗(yàn)樣品的生產(chǎn)規(guī)模:
由于口服固體制劑的生物利用度與生產(chǎn)規(guī)模密切相關(guān)����,故一般情況下應(yīng)不小于今后工業(yè)化生產(chǎn)規(guī)模的 1/10 或不小于 10 萬(wàn)個(gè)單位。
測(cè)定數(shù)量:
為達(dá)到統(tǒng)計(jì)學(xué)要求����,規(guī)定每個(gè)品種的測(cè)定皆應(yīng)選取 12 個(gè)樣本。但鑒于目前溶出儀尚未有 12 個(gè)溶出杯裝置�,且考慮到工作量等諸多因素,一般測(cè)定 6 個(gè)樣本即可����。
溶出度試驗(yàn)參數(shù)的確定
1 裝置與轉(zhuǎn)速的確定
對(duì)片劑,建議采用槳板法����、50 r/min;對(duì)膠囊劑�,建議采用轉(zhuǎn)籃法、50 r/min( 如采用槳板法����,則建議采用沉降籃 )�。
通常認(rèn)為 50 r/min 與中老年人體內(nèi)胃腸道蠕動(dòng)強(qiáng)度基本一致���。除非特指或特殊制劑�,不宜采用更慢轉(zhuǎn)速����。一般采用大杯法,介質(zhì)體積為 900~1 000 ml����,不建議采用小杯法。
如溶出樣品濃度過(guò)低����,可采用加大進(jìn)樣量、同時(shí)加大流動(dòng)相中有機(jī)相比例����,使被測(cè)組分峰保留時(shí)間縮短、峰形尖銳���,以提高檢測(cè)靈敏度�。
2 試驗(yàn)參數(shù)
在某溶出介質(zhì)中����,當(dāng)結(jié)束時(shí)間點(diǎn)溶出量仍達(dá)不到普通制劑 90%、緩控釋制劑 85%時(shí)����,則可酌情放寬溶出度試驗(yàn)參數(shù)。建議先增加轉(zhuǎn)速至 75 r/min����,如未果,則可在溶出介質(zhì)中添加表面活性劑����,添加濃度應(yīng)以 0.01% (w/v) 為起點(diǎn)、逐步增加�,不建議采用 1.0%以上濃 度。
如仍未達(dá)標(biāo)���,則再增加轉(zhuǎn)速至 100 r/min���,但絕不允許添加有機(jī)溶劑。因?yàn)檫@將嚴(yán)重背離體內(nèi)外相關(guān)性原則,同時(shí)大大降低區(qū)分效應(yīng)����。
添加表面活性劑時(shí),應(yīng)注意不同來(lái)源試劑可能會(huì)對(duì)試驗(yàn)結(jié)果帶來(lái)顯著性差異的情況����,尤其是在使用十二烷基磺酸鈉時(shí),故建議在質(zhì)量標(biāo)準(zhǔn)中注明試劑來(lái)源���,以使試驗(yàn)重現(xiàn)����。
溶出曲線的測(cè)定
1 測(cè)定法
采用紫外法測(cè)定時(shí)�,由于存在著輔料、膠囊殼等諸多因素的干擾����,尤其在短波長(zhǎng) ( 低于 230 nm) 處,建議采用兩點(diǎn)相減法�,即一波長(zhǎng)為被測(cè)組分最大吸收波長(zhǎng),另一波長(zhǎng)為遠(yuǎn)端無(wú)吸收波長(zhǎng)���。該法可大大降低輔料干擾�,并在日本橙皮書中廣泛使用。
當(dāng)以上方法無(wú)法排除測(cè)定干擾����,或吸光度低 于 0.25( 即使采用 5 cm 長(zhǎng)距離測(cè)定池 )���,建議采用 HPLC 法���。
無(wú)論采用何法測(cè)定,皆建議對(duì)照品溶液濃度配制成 50%~60%釋放量����,以兼顧溶出液的不同濃度,盡可能縮小外標(biāo)一點(diǎn)法測(cè)定誤差���。
2 樣品處理
采用一個(gè)濾膜即可���,可大大提高工作效率,最大程度排除濾膜吸附的干擾����。
按規(guī)定,溶出液取出后應(yīng)過(guò)濾�,但考慮到濾膜吸附的影響 ( 特別是經(jīng)微粉化處理的小規(guī)格難溶藥物制劑 )���,建議:
①如采用 HPLC 法測(cè)定,建議溶出液離心后���,取上清液測(cè)定�。甚至直接置液相小瓶中����,靜置后進(jìn)樣。這樣����,便可僅取溶出液 2 ml,該體積相對(duì)于整個(gè)溶出液 (900~1 000 ml) 體積的影響很小�,故可省略其后的結(jié)果累積校正。
②如采用 UV 法測(cè)定���,由于每一時(shí)間點(diǎn)均應(yīng)至少棄去 5 ml 初濾液���,故抽取體積應(yīng)不少于 10 ml。由于體積較大�, 則應(yīng)進(jìn)行結(jié)果累積校正,否則不利于準(zhǔn)確判斷����。
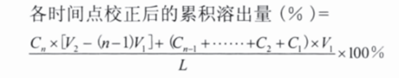
累積釋放度校正計(jì)算公式
多次取樣時(shí)�,可采取及時(shí)補(bǔ)充同體積同溫度溶出介質(zhì)���,亦可采取不補(bǔ)液兩種方式�,但必須保證每次抽取體積的固定性����。累積校正計(jì)算公式如下:
1 補(bǔ)液
其中 Cn 為各時(shí)間點(diǎn)取出后的樣品濃度 ( 即稀釋前的 )�;L 為制劑標(biāo)示量 (L/V2 單位需與 Cn 一致 );V1 為各時(shí)間點(diǎn)固定取樣體積�;V2 為溶出介質(zhì)體積。
該公式如采用各時(shí)間點(diǎn)測(cè)得釋放量表示�,則可演變?yōu)椋?/span>
其中 An 為各時(shí)間點(diǎn)測(cè)得溶出量。
2 不補(bǔ)液
計(jì)算時(shí)間點(diǎn)的確定
普通制劑如 15 min 內(nèi)溶出量達(dá) 85%以上����,則無(wú)需進(jìn)行曲線比較。此時(shí)����,仿制制劑亦應(yīng)滿足此條件。
只要 15 min 時(shí)溶出量未達(dá) 85%以上���,需進(jìn)行溶出曲線的比較�。
溶出量在 85% ( 緩控釋制劑 80%以上 ) 以上的時(shí)間點(diǎn)僅能選取一個(gè);且由于所采用的相似因子比較法 —— f2 因子法計(jì)算結(jié)果有依賴于比較時(shí)間 點(diǎn)個(gè)數(shù)的特性���,故建議選取溶出量間隔相近的 4~5 個(gè) ( 普通制劑 ) 或 4~6 個(gè) ( 緩控釋制劑 ) 時(shí)間點(diǎn)進(jìn)行計(jì)算比較���,但這些時(shí)間點(diǎn)的間隔無(wú)需相等。
如上文所列試驗(yàn)�,“當(dāng)結(jié)束時(shí)間點(diǎn)溶出量仍達(dá)不到普通制劑 90%、緩控釋制劑 85%時(shí)����,建議先增加轉(zhuǎn)速至 75 r/min,如未果���,則可在溶出介質(zhì)中添加表面活性劑���,如仍未達(dá)標(biāo),則再增加轉(zhuǎn)速至 100 r/min”����。
若最終溶出量仍未達(dá)到標(biāo)準(zhǔn),不建議再采用更為極端的條件�。
此時(shí)���,依據(jù)最終測(cè)定結(jié)果,選取溶出量間隔相近的 4 個(gè)時(shí)間點(diǎn)計(jì)算比較即可�。
對(duì)于所選時(shí)間點(diǎn)溶出結(jié)果變異系數(shù)的規(guī)定
所選用的第一時(shí)間點(diǎn)溶出結(jié)果變異系數(shù)應(yīng)不超過(guò) 20%,自第二時(shí)間點(diǎn)至最后時(shí)間點(diǎn)溶出結(jié)果變異系數(shù)均應(yīng)不得過(guò) 10%�。
若超出,應(yīng)從儀器的適用性予以考慮解決����,如增加轉(zhuǎn)速或增加表面活性劑濃度,直至滿足精密度要求�。
二���、測(cè)定仿制制劑
通過(guò)以上對(duì)參比制劑的測(cè)定����,確立“溶出曲線測(cè)定時(shí)間點(diǎn)”后�,在這些時(shí)間點(diǎn)分別測(cè)定仿制制劑。
仿制制劑的含量與參比制劑的差值應(yīng)在 5%以內(nèi)�,且所選用的樣品應(yīng)在重量 / 裝量差異所規(guī)定范圍的 1/2 以內(nèi),以盡可能排除因個(gè)體差異給溶出度試驗(yàn)數(shù)據(jù)帶來(lái)的影響���。
其生產(chǎn)規(guī)模亦應(yīng)同參比制劑����。
如所選時(shí)間點(diǎn)的溶出結(jié)果變異系數(shù)超出規(guī)定,已說(shuō)明該廠家產(chǎn)品批內(nèi)差異的不良波動(dòng)性�。此時(shí)則無(wú)需再進(jìn)行批間差異比較,但仍需計(jì)算出均值用于與參比制劑的比較���。
三�、溶出曲線比較法
目前����,日本和美國(guó)官方皆規(guī)定采用相似因子比較法 ——f1 因子和 f2 因子法進(jìn)行溶出曲線的比較。具體計(jì)算公式如下:
當(dāng)用于不同來(lái)源制劑間比較時(shí)����, ƒ2 因子應(yīng)≥ 50;當(dāng)用于同一來(lái)源制劑間比較時(shí) ( 批間 / 批內(nèi)差異�、各種變更等 ),ƒ2 因子應(yīng)≥ 65�。
表 7 為溶出量平均差異與相應(yīng) ƒ2 因子臨界值的關(guān)系。
由此可見����,若直觀評(píng)估、各時(shí)間點(diǎn)差異在 10%或 5%以內(nèi),則可基本斷定 ƒ2 因子大于 50 或大于 65���。
溶出曲線比較時(shí)����,有時(shí)會(huì)出現(xiàn)仿制制劑溶出曲線明顯高于 ( 或快于 ) 原研制劑�,這并不能說(shuō)明仿制制劑質(zhì)量?jī)?yōu)于原研制劑,生物利用度試驗(yàn)才能最終判斷兩者是否具有等效性���。
來(lái)源:《中國(guó)醫(yī)藥工業(yè)雜志》
作者:張啟明 �,謝沐風(fēng)���,寧保明 �,庾莉菊