任何影響藥物純度的物質(zhì)統(tǒng)稱為雜質(zhì)�����。藥品在臨床使用中產(chǎn)生的不良反應(yīng)除了與藥品本身的藥理活性有關(guān)外����,有時(shí)與藥品中存在的雜質(zhì)也有很大關(guān)系,如青霉素等抗生素中的多聚物等高分子雜質(zhì)就是引起過敏反應(yīng)的主要原因��。
因此雜質(zhì)研究是藥品研發(fā)的一項(xiàng)重要內(nèi)容�����。
《中華人民共和國(guó)藥典》(以下簡(jiǎn)稱《中國(guó)藥典 》) 2005年版附錄中收載了《藥品雜質(zhì)分析指導(dǎo)原則》��,原國(guó)家食品藥品監(jiān)督管理局也于 2005年頒布了《化學(xué)藥物雜質(zhì)研究的技術(shù)指導(dǎo)原則》 ��,其中概述了雜質(zhì)研究的內(nèi)容和思路�����。
筆者結(jié)合藥品審評(píng)實(shí)踐以及與部分國(guó)內(nèi)外制藥企業(yè)在雜質(zhì)研究實(shí)踐方面的交流����,談?wù)剛€(gè)人對(duì)化學(xué)藥品雜質(zhì)研究思路的理解和認(rèn)識(shí)。
一��、雜質(zhì)研究的總體原則
雜質(zhì)可能產(chǎn)生的來源為:工藝過程 (包括合成中未反應(yīng)完全的反應(yīng)物及試劑�����、中間體����、副產(chǎn)物以及反應(yīng)物及試劑中混入的雜質(zhì) )�����、降解過程,所以雜質(zhì)研究的總體原則就是要結(jié)合在研產(chǎn)品具體工藝以及產(chǎn)品特點(diǎn)開展研究��。
首先��,要結(jié)合具體工藝及產(chǎn)品特點(diǎn)來分析產(chǎn)品中可能產(chǎn)生何種雜質(zhì)����,通過雜質(zhì)譜的分析對(duì)產(chǎn)品中雜質(zhì)的來源及結(jié)構(gòu)情況有較為全面的了解;
然后��,在雜質(zhì)譜分析的基礎(chǔ)上��,有針對(duì)性地選擇合適的分析方法��,以確保雜質(zhì)的有效檢出及控制�����;
最后��,需綜合藥學(xué)、藥理毒理及臨床研究結(jié)果確定合理的雜質(zhì)限度�����,從而保證藥品的質(zhì)量及安全性��。
二��、雜質(zhì)譜的分析
1 基于合成路線進(jìn)行工藝雜質(zhì)的分析
對(duì)于原料藥�����,需依據(jù)所采用的具體合成工藝來分析在研產(chǎn)品中可能產(chǎn)生的雜質(zhì)�����。
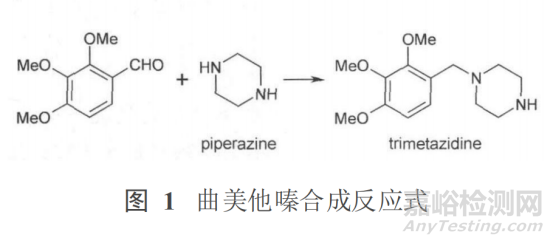
例如:抗心絞痛藥物鹽酸曲美他嗪質(zhì)量標(biāo)準(zhǔn)中哌嗪的檢查正是基于曲美他嗪的合成路線確定��。見圖 1����。
由圖 1可見,哌嗪是曲美他嗪的反應(yīng)物之一�����,可能會(huì)因未反應(yīng)完全而殘留在終產(chǎn)品中,是研究中需要關(guān)注的一個(gè)工藝雜質(zhì)����。
2 基于產(chǎn)品的結(jié)構(gòu)特征分析可能產(chǎn)生的降解產(chǎn)物
降解產(chǎn)物主要與藥物的結(jié)構(gòu)特征密切相關(guān)。
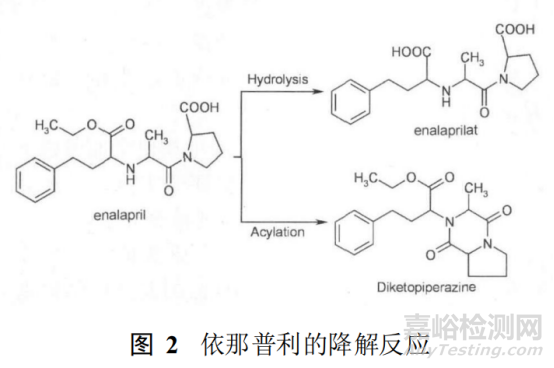
例如抗高血壓藥物依那普利�����,其結(jié)構(gòu)中含有羧酸乙酯基�����,該基團(tuán)易發(fā)生水解反應(yīng)��,生成羧基����,即產(chǎn)生依那普利拉�����;此外�����,依那普利的結(jié)構(gòu)中含有 1 個(gè)羧基,同時(shí)還含有 1個(gè)氨基����,它們易發(fā)生酰化反應(yīng)生成內(nèi)酰胺結(jié)構(gòu)����,即產(chǎn)生依那普利二酮哌嗪。見圖 2��。
故《中國(guó)藥典 》2005年版二部收載的馬來酸依那普利拉質(zhì)量標(biāo)準(zhǔn)中有關(guān)物質(zhì)項(xiàng)下控制依那普利拉和依那普利二酮哌嗪的檢查����。
通過結(jié)構(gòu)特點(diǎn)來分析可能的降解產(chǎn)物是一種預(yù)測(cè),另外還可以通過強(qiáng)制破壞試驗(yàn)來驗(yàn)證可能的降解產(chǎn)物和降解途徑�����。
通常國(guó)內(nèi)比較常見的是采用酸�����、堿����、氧化����、高溫��、光照破壞性試驗(yàn)來進(jìn)行分析方法專屬性的驗(yàn)證�����,而很少關(guān)注降解產(chǎn)物結(jié)構(gòu)的分析�����。
強(qiáng)制破壞試驗(yàn)不一定局限在以上羅列條件內(nèi)����,可以采取更靈活的方式來更好地預(yù)測(cè)產(chǎn)品可能的降解途徑和降解產(chǎn)物��。
例如對(duì)于原料藥����,可以考察溶液狀態(tài)下產(chǎn)品在破壞性條件下的變化。必要時(shí)��,可以進(jìn)行以上因素綜合存在時(shí)的強(qiáng)制降解試驗(yàn)。
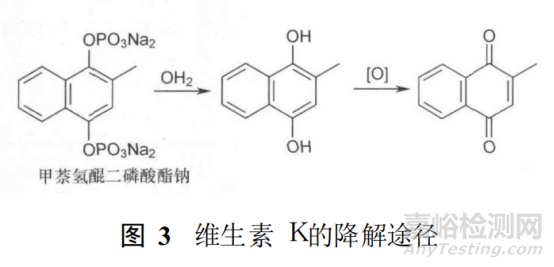
例如��,維生素 K的人工合成品甲萘氫醌二磷酸酯鈉�����,在一定 pH值水解條件下�����,首先生成甲萘氫醌����;進(jìn)而在氧化條件下,甲萘氫醌發(fā)生氧化反應(yīng)��,生成甲萘醌��,反應(yīng)過程見圖 3�����。
除此�����,破壞性試驗(yàn)中需關(guān)注破壞條件不宜過于劇烈,以免引起二次降解��。
在進(jìn)行了破壞性試驗(yàn)后����,應(yīng)結(jié)合藥物特點(diǎn)和必要的結(jié)構(gòu)分析手段,對(duì)潛在的降解產(chǎn)物進(jìn)行結(jié)構(gòu)確認(rèn)��,以便于下一步分析方法的研究驗(yàn)證以及限度制定��。
3 基于全面的研究數(shù)據(jù)分析產(chǎn)品中最終可能存在的雜質(zhì)
以上研究是分析和預(yù)測(cè)產(chǎn)品中可能存在的雜質(zhì)����,但在產(chǎn)品實(shí)際的生產(chǎn)過程和貯存包裝條件下,并不一定產(chǎn)生上述所有的雜質(zhì)�����。
故最終對(duì)產(chǎn)品的控制����,尚需結(jié)合樣品在生產(chǎn)過程中實(shí)際產(chǎn)生的雜質(zhì)情況以及在加速和長(zhǎng)期留樣穩(wěn)定性研究中實(shí)際產(chǎn)生的雜質(zhì)情況來確定����。
三、雜質(zhì)分析方法的研究
分析方法的選擇直接關(guān)系到雜質(zhì)測(cè)定結(jié)果的專屬性與準(zhǔn)確性。
不同的檢測(cè)方法可能獲得不同的檢測(cè)結(jié)果����,雜質(zhì)檢查分析方法建立的根本原則是應(yīng)專屬、靈敏����,主成分與雜質(zhì)能達(dá)到良好分離,檢測(cè)限能滿足限度檢查的需求����。
關(guān)于雜質(zhì)分析方法的研究與驗(yàn)證在已發(fā)布的指導(dǎo)原則中有詳細(xì)描述,此處重點(diǎn)針對(duì)目前注冊(cè)研究中存在的主要問題進(jìn)行分析��。
1 分離方法的研究
在對(duì)雜質(zhì)譜進(jìn)行分析的基礎(chǔ)上����,方法的建立及驗(yàn)證就具有較強(qiáng)的針對(duì)性。
由于各種分析方法均具有一定的局限性����,研究過程中,需關(guān)注不同原理的分析方法間的相互補(bǔ)充與驗(yàn)證�����。
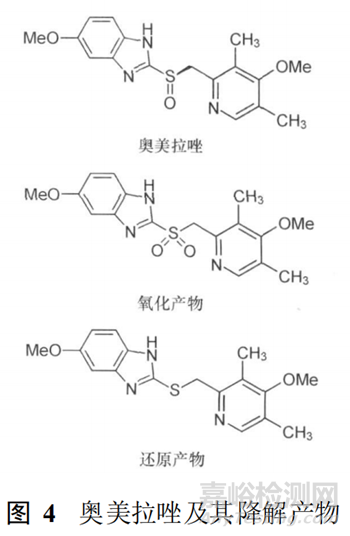
如 《中國(guó)藥典》收載的奧美拉唑質(zhì)量標(biāo)準(zhǔn)中采用 HPLC檢測(cè)方法于 280 nm進(jìn)行有關(guān)物質(zhì)的測(cè)定,分析該檢測(cè)條件主要是檢測(cè)奧美拉唑的氧化產(chǎn)物奧美拉唑磺?�;?���,但英國(guó)藥典標(biāo)準(zhǔn)在此基礎(chǔ)上專門列出了 TLC方法進(jìn)行奧美拉唑還原產(chǎn)物的測(cè)定。
注冊(cè)申報(bào)中����,最常見的一個(gè)現(xiàn)象是同品種國(guó)外藥典采用梯度洗脫方法,而自研品種在無充分研究的基礎(chǔ)上采用等度洗脫����。梯度洗脫確實(shí)存在基線漂移大,操作相對(duì)繁瑣的缺點(diǎn)����,但等度洗脫具有可能漏檢以及費(fèi)時(shí)的缺點(diǎn)。
故在進(jìn)行方法變更的過程中����,需要對(duì)變更前后方法的檢測(cè)能力進(jìn)行充分的對(duì)比研究�����。
另外,在進(jìn)行分析方法專屬性驗(yàn)證的過程中��,建 議對(duì)主成分的峰純度進(jìn)行檢查��,以考證和主成分結(jié)構(gòu)類似的雜質(zhì)是否因分離不完全而涵蓋在主成分色譜峰中��。
2 定量方法的選擇
雜質(zhì)常規(guī)的定量方法包括雜質(zhì)對(duì)照品法����,加校正因子的主成分自身對(duì)照法,不加校正因子的主成分對(duì)照法以及面積歸一化法����。
從目前藥品的研究注冊(cè)情況分析,不加校正因子的主成分對(duì)照法是較常采用方法�����。
因雜質(zhì)同主成分響應(yīng)因子可能不同��,故直接采用不加校正因子的主成分對(duì)照法缺乏科學(xué)性�����。
理想的定量方法為已知雜質(zhì)對(duì)照品法與未知雜質(zhì)不加校正因子的主成分自身對(duì)照法兩者的結(jié)合����。
當(dāng)然����,如果研究證明已知雜質(zhì)和主 成分的相對(duì)響應(yīng)因子在 0.9~1.1的范圍內(nèi)�����,可以用主成分自身對(duì)照法計(jì)算含量����,例如美國(guó)藥典收載的紫杉醇質(zhì)量標(biāo)準(zhǔn)中多個(gè)已知雜質(zhì)的控制均采用主成分自身對(duì)照法。
四��、雜質(zhì)限度的確定
雜質(zhì)限度確定的總體原則是在可行的范圍內(nèi)盡可能低��。
影響雜質(zhì)限度確定的因素有:雜質(zhì)的安全范圍����、藥品實(shí)際的生產(chǎn)能力、穩(wěn)定性期間的變化程度��、檢測(cè)方法的變異性����。
對(duì)于不同注冊(cè)類別的藥品而言����,因可參考信息量的不同����,雜質(zhì)限度的確定思路會(huì)有些不同����。
1 創(chuàng)新藥雜質(zhì)限度的確定思路
創(chuàng)新藥的研究是一個(gè)逐漸認(rèn)知的過程,其間可能會(huì)經(jīng)歷處方工藝的調(diào)整�����、雜質(zhì)譜的改變�����、分析方法的優(yōu)化等變化����,對(duì)于雜質(zhì)的研究和認(rèn)識(shí)是一個(gè)不斷積累的過程,故雜質(zhì)的限度確定也具有階段性�����。
從申報(bào)臨床的角度而言,雜質(zhì)限度確定的主要原則是保證臨床用樣品的安全性��,雜質(zhì)限度確定的依據(jù)主要是已進(jìn)行的臨床前安全性研究中獲得的結(jié)果�����,通常是要求用于臨床試驗(yàn)的樣品雜質(zhì)不得超過用于臨床前安全性研究的樣品��。
從申報(bào)上市的角度而言��,雜質(zhì)限度的確定就要在已有的基礎(chǔ)上����,結(jié)合生產(chǎn)工藝的放大和優(yōu)化,多批產(chǎn)品生產(chǎn)數(shù)據(jù)以及穩(wěn)定性信息�����,本著盡可能低的原則制定合理限度����。
通常從申報(bào)臨床到申報(bào)上市,對(duì)于雜質(zhì)的研究會(huì)越來越充分��,對(duì)于雜質(zhì)的控制越來越嚴(yán)格。
例如��,某藥品在申報(bào)臨床階段�����,最大單個(gè)雜質(zhì)控制為不得超過 1.0%��,總雜質(zhì)控制為不得超過 3.0%����;申報(bào)上市階段��,控制工藝雜質(zhì) A不得超過 0.25%��,降解產(chǎn)物 B 不得超過 0.25%��,其他單個(gè)最大雜質(zhì)不得超過 0.15%�����,總雜質(zhì)不得超過 1.25%����。
從申報(bào)臨床到申報(bào)上市,產(chǎn)品總雜質(zhì)限度趨于嚴(yán)格,并且對(duì)于單個(gè)已知雜質(zhì)進(jìn)行了定性和定量研究����,分別制定限度。
為了說明雜質(zhì)限度確定過程中如何全面考慮上述提及的各因素�����,也列舉一個(gè)審評(píng)的實(shí)例����。
某產(chǎn)品中雜質(zhì) C為其降解產(chǎn)物,可獲得的信息如下����。從安全性研究結(jié)果分析,當(dāng)雜質(zhì)含量在 3.57% 時(shí)獲得的安全性結(jié)果與未破壞樣品的安全性結(jié)果基本無差異�����,說明雜質(zhì) C含量在 3.57%時(shí)為可接受的安全限度����。
從生產(chǎn)和貯存結(jié)果分析,32批產(chǎn)品中雜質(zhì) C的實(shí)測(cè)范圍為 0.04%~ 0.87%�����,平均含量為 0.38%,標(biāo)準(zhǔn)差為 0.21��,平均值 ±3 ×標(biāo)準(zhǔn)偏差為 1.0%����。計(jì)算限度 1.0%符合生產(chǎn)的波動(dòng)范圍,同時(shí)也低于安全性限度����,故可做為雜質(zhì) C控制的合理限度����。
2 仿制藥雜質(zhì)限度的確定思路
對(duì)于仿制產(chǎn)品而言,相比于創(chuàng)新藥��,雜質(zhì)安全性信息的獲取來源可能不同����。
一則可以自行進(jìn)行雜質(zhì)的安全性研究,二則可以參考已上市產(chǎn)品的安全性信息��。
從研究的周期和研究投入角度而言����,大部分研發(fā)者會(huì)選擇后一種方式��,那就需要將自研產(chǎn)品和被仿制產(chǎn)品進(jìn)行全面的雜質(zhì)對(duì)比研究��,包括雜質(zhì)的種類��、雜質(zhì)的含量��。如無特殊理由�����,自研產(chǎn)品所定雜質(zhì)限度不得超過被仿產(chǎn)品的雜質(zhì)限度�����。
下面列舉一個(gè)仿制注射劑產(chǎn)品雜質(zhì)研究的實(shí)例來做具體分析����,表 1中列出了雜質(zhì)研究中獲得的對(duì)比數(shù)據(jù)��。
仿制品中 3個(gè)已知雜質(zhì) (A�����,B,D)高于上市品����,且高于 ICH規(guī)定的可接受限度 0.1%;此外��,兩個(gè)未 知雜質(zhì) (RRT1.28和 1.31)也高于 0.1%�����。
關(guān)于雜質(zhì) A����,缺乏試驗(yàn)和文獻(xiàn)資料支持其安全性,故應(yīng)改善工藝����,降低其含量��,使之不超過上市產(chǎn)品的限度����;
關(guān)于雜質(zhì) B,為一上市藥物��,有長(zhǎng)期人用歷史,文獻(xiàn)資料顯示其在體內(nèi)可轉(zhuǎn)化成主藥�����,不存在安全性方面的擔(dān)心��,可接受目前的雜質(zhì)水平��;
關(guān)于雜質(zhì) D�����,文獻(xiàn)顯示其為主藥在人體的主要代謝產(chǎn)物�����,也不存在安全性擔(dān)心�����;
關(guān)于 RRT1.28和 1.31的未知雜質(zhì)����,因缺乏試驗(yàn)和文獻(xiàn)資料支持其安全性,應(yīng)改善工藝將其降低到未知雜質(zhì)可接受的 0.1%限度以下�����。
雜質(zhì)研究是藥品研究的一項(xiàng)重要內(nèi)容,貫穿于藥品研發(fā)的始終����。伴隨著對(duì)藥品研發(fā)規(guī)律的認(rèn)知、 質(zhì)量源于設(shè)計(jì)理念的不斷拓展及新技術(shù)����、新方法的不斷涌現(xiàn),雜質(zhì)研究必將會(huì)有新的突破��。