1����、 安全性評價樣品的重要性
創(chuàng)新藥研發(fā)是通過疾病靶點(diǎn)研究、結(jié)構(gòu)篩選��、工藝設(shè)計(jì)與優(yōu)化(或處方與工藝設(shè)計(jì))����、結(jié)構(gòu)確證、質(zhì)量控制�、穩(wěn)定性研究、藥效與藥代研究�����、安全性評價(毒理研究)等一系列研究,將一個全新分子投入到市場用于治療特定疾病的復(fù)雜過程��。這個過程中��,安全性評價是不可或缺的重要環(huán)節(jié)��,也是新藥研發(fā)終止的重要原因之一�,是創(chuàng)新藥研發(fā)永恒關(guān)注的問題。
臨床前安全性評價試驗(yàn)樣品包括原料藥安全性評價樣品及制劑安全性評價樣品�����,分別評價新化學(xué)實(shí)體及新制劑的安全性���。
臨床前安全性評價主要是動物毒理試驗(yàn),是創(chuàng)新藥從動物研究到人體研究的橋梁���,通過動物毒理試驗(yàn)①可以初步判定藥物的毒副作用���,為開展臨床提供安全性數(shù)據(jù)支持。②可以為雜質(zhì)限度制定提供依據(jù)���;③依據(jù)試驗(yàn)結(jié)果��,在安全性可支持的范圍內(nèi)雜質(zhì)水平可適當(dāng)放寬��,可以一定程度上降低臨床試驗(yàn)樣品的生產(chǎn)壓力���,不必花費(fèi)大量時間過度優(yōu)化工藝���,為項(xiàng)目推進(jìn)爭取時間;④如在毒理試驗(yàn)中發(fā)現(xiàn)不可接受的毒性可及時止損��;⑤在后期臨床研究中如出現(xiàn)不良反應(yīng)���,臨床前毒理數(shù)據(jù)可為原因調(diào)查提供依據(jù)�;⑥毒理試驗(yàn)結(jié)果可為制劑開發(fā)規(guī)格制定提供重要支撐�。
2、 安全性評價樣品雜質(zhì)種類和水平
2.1雜質(zhì)存在的意義
安全評價樣品雜質(zhì)譜的控制至關(guān)重要�。正常情況下,產(chǎn)品工藝開發(fā)與優(yōu)化過程是追求雜質(zhì)最小化的過程���,樣品純度越高�,安全性越好���;而在毒理研究中���,樣品純度不是追求的目標(biāo)���,因?yàn)槎纠順悠分械碾s質(zhì)肩負(fù)著雜質(zhì)安全性評價的重任,只有存在于毒理樣品中才能獲得評價���,積累安全性數(shù)據(jù)���。
2.2雜質(zhì)種類
那么毒理樣品中應(yīng)含有哪些雜質(zhì)呢?(以下均為個人觀點(diǎn))
有機(jī)雜質(zhì)
(1)有關(guān)物質(zhì)
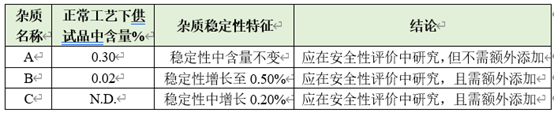
l在穩(wěn)定工藝下���,多批樣品中均存在���,且原料藥中含量超過0.1%(或制劑中含量超過0.15%)的雜質(zhì)(工藝雜質(zhì))����,如下表中雜質(zhì)A。
l樣品中存在�����,含量較低,但在穩(wěn)定性考察中含量顯著增長的雜質(zhì)(工藝雜質(zhì)&降解雜質(zhì))����,如下表中雜質(zhì)B。
l樣品中不存在���,但在穩(wěn)定性考察中新增產(chǎn)生的雜質(zhì)(降解雜質(zhì))�����,如下表中雜質(zhì)C�����。
(2)異構(gòu)體
異構(gòu)體與API結(jié)構(gòu)相同���,但藥效、藥代���、毒性可能具有顯著差異�����,是產(chǎn)生毒副作用風(fēng)險來源之一��,F(xiàn)DA要求對手性藥物進(jìn)行毒理研究時���,應(yīng)獲得各單一構(gòu)型的立體異構(gòu)體���,進(jìn)行必要的比較研究,以確定擬進(jìn)一步開發(fā)的藥物�����。從這一條規(guī)定看�����,立體異構(gòu)體在確定候選藥物的時候已需毒理研究���,理論上在臨床前安全性評價時��,可以不再重復(fù)考察��,但為進(jìn)一步確認(rèn)異構(gòu)體的安全限度,可以選用異構(gòu)體含量稍高的樣品或添加適當(dāng)濃度異構(gòu)體進(jìn)行安全性評價研究�����。
(3)遺傳毒雜質(zhì)
終產(chǎn)品中遺傳毒雜質(zhì)一定會被嚴(yán)格控制在安全限度之下,不會產(chǎn)生難以預(yù)料或不可接受的毒性��,因此臨床前安全性評價樣品中通常不需要額外添加遺傳毒雜質(zhì)����。如確實(shí)需考察其安全性應(yīng)通過定向合成單獨(dú)考察其毒性���。
(4)聚合物
聚合物與有關(guān)物質(zhì)的研究策略基本一致��,如果聚合物含量較高或穩(wěn)定性中明顯增長的話需要額外添加考察其安全性�����。
無機(jī)雜質(zhì)
通常擁有充分的安全性研究資料并具有確定的限度(ICH Q3D)��,一般無需額外添加考察安全性。
殘留溶劑
通常擁有充分的安全性研究資料并具有確定的限度(ICH Q3C)�,一般無需額外添加考察安全性。
2.3雜質(zhì)水平
安全性評價樣品中雜質(zhì)水平是另一個需要重點(diǎn)考察的因素����,也是一個難度系數(shù)超高的技術(shù)活��。雜質(zhì)水平過高�,可能影響API毒理評價結(jié)果�����,造成安全性評價試驗(yàn)失?��?���;雜質(zhì)水平過低�����,其所能支持的雜質(zhì)安全水平有限�����,可能給后面臨床批樣品生產(chǎn)造成壓力���,耽誤研發(fā)進(jìn)程�。其中的風(fēng)險與獲益需要仔細(xì)評估���。
雜質(zhì)添加水平是沒有硬性規(guī)定的�����,個人認(rèn)為雜質(zhì)水平可以根據(jù)早期有限的毒性研究數(shù)據(jù)�����,結(jié)合目前的工藝水平和穩(wěn)定性來制定,也就是根據(jù)預(yù)期的毒理給藥水平����、目前工藝下樣品中雜質(zhì)的水平及穩(wěn)定性,倒推出毒理樣品中的雜質(zhì)水平���。例如�����,基于早期研究預(yù)期大鼠長毒Noael不會低于20 mg/kg/d���,臨床擬定的給藥劑量為120 mg/d�����,目前工藝下樣品中雜質(zhì)含量為0.6%���,那么倒推出安全性評價樣品中該雜質(zhì)的水平應(yīng)為=[(120 mg/kg/d 60 kg)0.6%37 kg/m2]/(20 mg/kg/d6 kg/m2)=0.37%。
2.4全性評價數(shù)據(jù)在早期雜質(zhì)限度制定中的應(yīng)用示例
CDE要求臨床批樣品中雜質(zhì)水平不得超出動物安全性試驗(yàn)數(shù)據(jù)所支持的相應(yīng)雜質(zhì)的水平�����。那么�,安全性評價樣品所能支持的水平怎樣計(jì)算呢?尤其是早期的IND階段可以怎樣計(jì)算呢���?
計(jì)算的方式有很多種���,核心原理是動物Noael所代表的暴露量的與人體之間的轉(zhuǎn)化。下面借鑒了文獻(xiàn)中的兩種計(jì)算方式:
這里需要說明的是���,以上的這些依據(jù)安全性評價結(jié)果推算雜質(zhì)限度的方法僅適用于研發(fā)初期���,因?yàn)榇藭r原料藥���、制劑工藝均尚未完全確定,雜質(zhì)限度可根據(jù)安全性評價結(jié)果制定����,只要確保臨床試驗(yàn)安全即可����,到了研發(fā)中后期,原料藥��、制劑工藝均尚基本確定����,雜質(zhì)限度原則上應(yīng)符合ICH要求。
3�����、安全性評價樣品制備方法
安全性評價用樣品的制備方法有以下幾種:
(1)雜質(zhì)可獲得情況下
u選擇一批雜質(zhì)種類相對較全的原料藥產(chǎn)品���,向其中添加一定水平的需要重點(diǎn)評估的雜質(zhì)(因?yàn)槭枪腆w混合��,因此要特別注意混合均勻性)�����。
u在反應(yīng)過程中加入一定水平的雜質(zhì)(需要提前考察各雜質(zhì)在反應(yīng)中的清楚和穩(wěn)定性)
(2)雜質(zhì)不可獲得的情況下
u對正常生產(chǎn)所獲得的產(chǎn)品進(jìn)行適當(dāng)?shù)慕到馄茐?��,獲得所需的雜質(zhì)水平
u通過調(diào)整工藝中某些參數(shù)獲得較高雜質(zhì)水平的樣品���。
4、安全性評價樣品提供時間
安全性評價樣品在什么時候提供比較適宜呢��?首先看下安全性評價試驗(yàn)的特點(diǎn)和審評要求:
①臨床前安全性評價試驗(yàn)時間跨度較長�����;
②部分研究需得到安全性評價結(jié)果(或進(jìn)行中的部分結(jié)果)的支持才能開展�����;
③臨床前安全性評價試驗(yàn)是IND申報的審評重點(diǎn)���,直接關(guān)系是否可進(jìn)一步開展臨床研究�,故CDE要求必須完成臨床前安全性評價才能申報���。
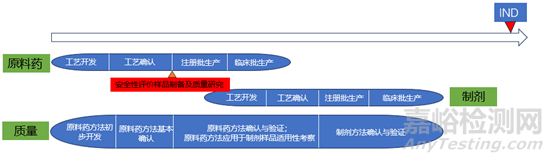
綜合以上幾點(diǎn)��,在實(shí)際研究過程中��,安全性評價試驗(yàn)常常是申報的限速步驟����,因此在工藝確認(rèn)之后應(yīng)盡快生產(chǎn)安全性評價樣品,盡早開展安全性評價試驗(yàn)(見下圖)���。
5�、分析方法
理論上用于安全評價用樣品的檢測方法最好是基本確認(rèn)的方法,方便雜質(zhì)的比對����,否則���,如果檢測方法后期發(fā)生較大改動�����,需要橋接兩個方法����,將雜質(zhì)一一比對,不要造成雜質(zhì)譜分析混淆��。
6����、穩(wěn)定性研究
應(yīng)對臨床前安全性評價樣品進(jìn)行穩(wěn)定性考察��,對影響安全性���、有效性的重要指標(biāo)進(jìn)行檢測,如外觀��、雜質(zhì)水平���、主成分含量���、晶型等�����。對臨床前安全性評價樣品進(jìn)行穩(wěn)定性研究��,一是要確保安全性評價期間樣品質(zhì)量可控,二是為臨床前安全性評價樣品的運(yùn)輸和儲存條件的選擇提供依據(jù)�����。
7�、總結(jié)
在新藥研發(fā)中,安全性評價樣品是較為特殊和重要的存在��,作為新藥研發(fā)人員,我們應(yīng)充分理解非臨床安全性評價的相關(guān)要求�����,充分掌握非臨床安全性評價對樣品的質(zhì)量要求�;充分利用非臨床安全性評價結(jié)果在質(zhì)量研究中的支撐作用��,助力新藥更快����、更穩(wěn)向前推進(jìn)����。
參考文獻(xiàn)
[1] 化學(xué)藥物雜質(zhì)研究技術(shù)指導(dǎo)原則
[2] 黃曉龍,對創(chuàng)新藥研發(fā)中雜質(zhì)限度確定的幾點(diǎn)思考,中國新藥雜質(zhì) 2007,16(2)��,97-100����。
[3]何伍,王海學(xué),淺談藥物雜質(zhì)限度的制訂方法���,中國醫(yī)藥工業(yè)雜志���,2009,40(10)�����,787-790�����。
[4]手性藥物研究指導(dǎo)原則