藥物的多晶型是影響藥物安全性和有效性的重要因素。多晶型藥物的物理化學性質差異而可能導致藥效的不同����,這些不同一般表現(xiàn)在晶型的外觀、溶解速率����、溶解度����、熔點����、光學和電子學性質、蒸汽壓和密度����、溶出度����、生物有效性等方面����,從而影響了藥物的穩(wěn)定性����、生物利用度及療效����。近些年來����,藥物的多晶型研究越來越受到醫(yī)藥研發(fā)與國家藥監(jiān)監(jiān)管領域的關注與重視。在ICH Q6A文件決策樹4(#1)~(#3)及2020版藥典《藥物晶型研究及晶型質量控制指導原則》均提供了與多晶型轉化相關的指導原則����,供研發(fā)人員參考借鑒����。接下來我們就結合相關指導原則,再結合文獻案例實際相關的研究工作情況深入了解指導原則的深刻意義����。因本人經驗不足,如有不當之處����,還請指出����,共同進步。
中國藥典指出:當固體藥物存在多晶型現(xiàn)象����,且不同晶型狀態(tài)對藥品的有效性����、安全性或質量可產生影響時����,應對原料藥物����、固體制劑、半固體制劑、混懸劑等中的藥用晶型物質狀態(tài)進行定性或定量控制����。藥品的藥用晶型應選擇優(yōu)勢晶型,并保持制劑中晶型狀態(tài)為優(yōu)勢晶型����,以保證藥品的有效性����、安全性與質量可控。但是要如何始終如一的保持優(yōu)勢晶型����,則需要研發(fā)人員要充分了解評估晶型轉化是否存在風險����,或者某種程度的轉化是否可以接受,需要重點考慮以下因素����,包括: 1)對晶型轉化機理的深刻理解����,從而可以制定控制策略����;2)檢測和量化晶型轉化的能力; 3)預測和測試晶型轉化對安全性和有效性的影響的能力����,以證明晶型控制策略的合理性����。
1.晶型轉化機理的深刻研究與理解
1.1多晶型篩選及轉化關系研究
對所選的原料藥的目標晶型要充分進行轉化風險評估����,以為下游制劑的開發(fā)奠定晶型研究的基礎����。做項目開發(fā)的最早期,盡可能進行徹底����、可靠的晶型篩選����,篩選的目的是盡可能地找到更多的晶型,并確定最適合����、穩(wěn)定的晶型進行研究����。多晶型篩選常用化學方法主要包括:重結晶法、快速溶劑去除法����、沉淀法����、種晶法等;常用物理方法主要包括:熔融結晶法����、晶格物理破壞法����、物理轉晶法等����。各種方法影響晶型物質形成的重要技術參數(shù)包括:溶劑(類型����、組成����、配比等)����、濃度、成核速率����、生長速率����、溫度����、濕度����、光度、壓力����、粒度等。如參考案例:達比加群酯甲磺酸鹽的多晶型研究主要是通過冷卻結晶法����,考察溶劑種類����、溶劑比例、析晶溫度等條件對達比加群酯甲磺酸鹽晶型的影響����,制備出幾種不同晶型的達比加群酯甲磺酸鹽晶體����。在開發(fā)過程中����,他們以無水形式I進行了開發(fā)����。
1.2 多晶型的表征方法
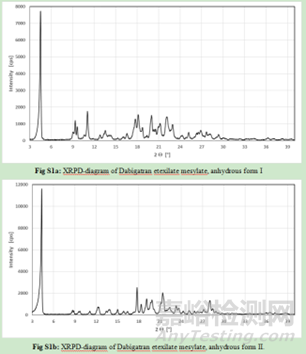
由于藥物多晶型現(xiàn)象����,快速有效并且準確的鑒別方法對多晶型的研究與表征很有必要����。目前,多晶型研究常用的表征方法有X射線衍射方法����、光譜分析方法、熱分析方法以及熔點分析法等����。近年來����,固態(tài)核磁、拉曼光譜����、近紅外光譜和漫反射紅外光譜也被廣泛的應用于藥物的晶型分析。
1.3 晶型可開發(fā)性評估
晶型可開發(fā)性評估一般需要根據(jù)不同晶型的特點����、種類和復雜性����,采用多種表征手段聯(lián)用的方式����,以提高對晶型理化性質的全面深入理解����。需要結合晶型方法的可加工性/穩(wěn)健性/可放大性、晶型的穩(wěn)定性����、制劑過程中的輔料相容性����、制劑穩(wěn)定性、溶出速率等方面進行充分評估確認,最終目的是選擇一種優(yōu)勢晶型����,以指導后續(xù)制劑處方工藝開發(fā)����,并形成控制策略。
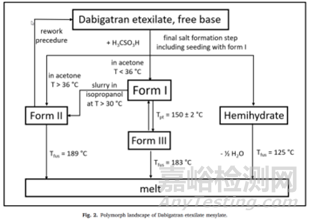
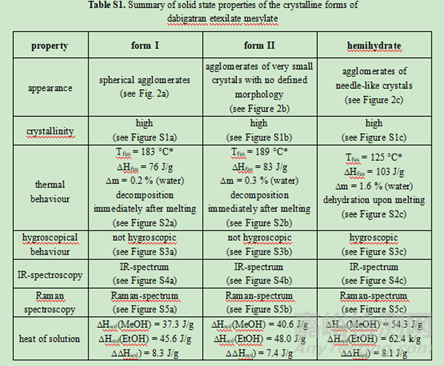
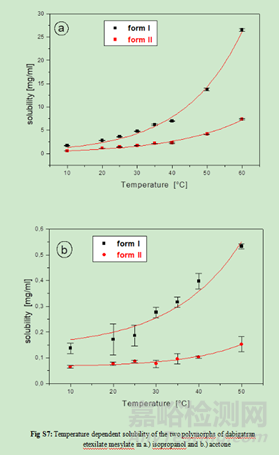
如達比加群酯甲磺酸鹽選擇無水物Ⅰ晶型作為目標晶型開發(fā)原因如下:1)原料藥生產中具有穩(wěn)健的可加工性:在鹽沉淀步驟中更容易分離出形式I晶型����。且形式I結晶通常呈球形團塊,具有良好的整體物理特性����,如易于過濾、易于干燥和優(yōu)異的流動特性����;2)它表現(xiàn)出稍好的長期化學穩(wěn)定性����。化學穩(wěn)定性數(shù)據(jù)表明����,無水形式I的化學穩(wěn)定性略高于無水形式II����。在36個月的時間里����,形式I的總雜質含量在25℃/60% RH.的條件下從0.30%增加到0.44%����,在40℃/75% RH的條件下從0.29%增加到0.98%����。形式II的總雜質含量在36個月內從25℃/60%RH的條件下從0.25%增加到0.85%����、40℃/75%RH保存時0.3% ~ 2.3%。半水化合物在熱力學上也是一種穩(wěn)定的形態(tài)����,但由于晶體形態(tài)不利����,沒有被選中進行開發(fā)����。半水結晶成很長很細的針狀物(見圖2c)����,導致比無水形式II更差的物理化學性質����。此外����,達比加群酯甲磺酸鹽對水解的敏感性阻止了其水合形式的發(fā)展。因此����,半水合物雖然熱力學穩(wěn)定����,但不能被選做目標晶型。
2.檢測和量化晶型轉化的能力
2.1晶型定性����、定量方法的開發(fā)
一般來說,特異性����、靈敏度和穩(wěn)健性是影響用于分析檢測技術決策的三個主要因素����。對于不同藥物的不同晶型,其檢查方法的專屬性是不同的����。進行晶型質量研究時����,應根據(jù)化合物的自身特點����,選擇適宜的����、具有專屬性的晶型檢查方法����?���;诰w學����、顯微鏡學、熱分析或光譜學的物理技術可用于表征晶體形式����。在早期開發(fā)過程中����,為了支持風險評估研究,重要的是開發(fā)一種適宜的分析方法����,該方法對晶體形式的變化具有廣泛的特異性����,但對輔料����、制粒工藝等的變化具有穩(wěn)健性����。每種技術檢測低水平晶型變化的能力取決于分子結構����、藥物負載、賦形劑種類等����,因此,無法直接比較或假設這些技術的靈敏度����,還是需要根據(jù)各個品種的自身特點去做詳細的分析檢測已開發(fā)出專屬性����、靈敏度適宜的檢測方法����。如下表為文獻中介紹的幾種固態(tài)表征技術開發(fā)時需要考慮的方法的專屬性����,以及對每個屬性效用的介紹����。
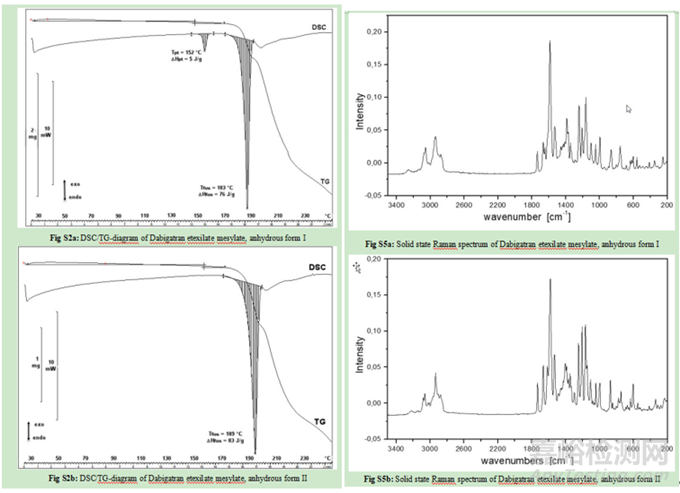
如可參考達比加群酯甲磺酸鹽的晶型I和晶型II定量分析的分析方法開發(fā)過程研究:
X射線粉末衍射和紅外光譜中兩種晶型形式的峰或信號強烈重疊����,因此無法考慮該兩種方法進行定量分析����。幸運的是����,DSC和拉曼光譜對該藥物的晶型檢測更為靈敏。因此開發(fā)了一種DSC方法����,能夠定量由晶型I組成的原料藥樣品中摻雜0.6%的晶型II����。但遺憾的是����,由于受輔料的干擾����,該DSC方法不能用于分析藥物成品及其制粒中間體����。,拉曼光譜能夠檢測顆粒制劑中的晶型II����,定量限為5%����。
2.2 晶型定量方法的實際應用評估
2.2.1 原料或制劑生產過程中的轉晶風險評估
利用2.1章節(jié)所開發(fā)的晶型定量方法����,需要應用該方法對多晶型藥物制劑進行晶型物質狀態(tài)的穩(wěn)定性研究����。在制劑的整個制備流程中,亞穩(wěn)態(tài)藥物晶型在受到溶劑����、溫度、濕度����、壓力����、研磨、輔料等影響時����,很容易發(fā)生藥物晶型的轉變。因此需要對原料����、制劑的各工藝進程進行轉晶風險評估����,研究內容包括:原料藥成分的晶型物質狀態(tài)的穩(wěn)定性、原料藥晶型物質與制劑處方中各種輔料的相容性����、制劑的制粒、成型����、干燥等工藝對原料藥晶型物質狀態(tài)的影響等。通過晶型物質狀態(tài)的穩(wěn)定性研究����,可為優(yōu)勢藥物晶型物質狀態(tài)選擇����、藥物制劑處方、制備工藝過程控制����、藥品貯存條件等提供科學依據(jù)����。
可參考達比加群酯甲磺酸鹽的晶型研究控制:
(1)原料藥合成
一系列實驗調查了晶型II是否可以在擬定的商業(yè)原料藥合成所描述的條件下或稍微偏離該擬定過程的條件下自發(fā)形成����。研究主要集中在合成成鹽步驟的兩個主要參數(shù):溫度和處理時間����。從實驗數(shù)據(jù)中有跡象表明����,轉變?yōu)榫虸I的風險隨著溫度和/或處理時間的延長而增加����。然而����,數(shù)據(jù)也表明����,在原料藥合成的最后一步����,當溫度不超過36℃時����,晶型I的可重復性沉淀是可行的����。第二組實驗研究了當存在晶型II種子時晶型I合成的穩(wěn)健性。從數(shù)據(jù)來看����,很明顯����,在幾個小時內就可以將晶型Ⅰ轉化為晶型Ⅱ。為了避免這種轉化����,應避免與晶型II種子接觸。綜上所述����,為避免在原料藥合成過程中形成熱力學更穩(wěn)定的II型����,在擬定的商業(yè)化工藝中實施了以下措施:鹽沉淀步驟的溫度已降至36℃以下����,其他操作條件也相應優(yōu)化,例如����,在原料藥分離步驟中,干燥器在低于50°C的溫度下使用����。
(2)原料藥受機械應力的影響研究
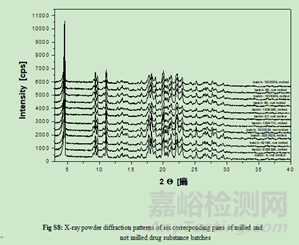
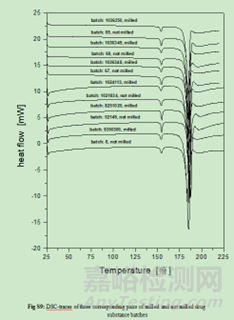
在銑削或壓縮等機械應力的影響下����,無水形式I向無水形式II的轉變也可能發(fā)生。對于甲磺酸達比加群酯����,在結晶不能直接滿足指定粒度分布的情況下,研磨是一個可選的工藝步驟����。因此,銑削必須被視為另一種可能的來源����,以形成熱力學上更穩(wěn)定的無水晶型II����。另一方面,通過壓縮向無水晶型II的轉化可以忽略不計����,因為在達比加群酯膠囊的制造過程中不包括壓縮步驟����,是將原料藥直接噴在酒石酸丸上。
主要通過X射線粉末衍射����、熱分析和溶液量熱(溶液熱)等手段����,對不同批次的研磨和未研磨原料藥批次進行分析����,以觀察研磨過程是否引起固態(tài)相變(部分或全部轉變?yōu)闊o水型II或非晶態(tài))����。結果顯示所有方法(原則上能夠檢測固態(tài)相變)都沒有檢測到研磨和未研磨之間的任何差異����,因此可以得出結論,沒有發(fā)生向無水型II (LOQ: 0.6%)或非晶態(tài)的轉變����。
(3)制劑生產過程中轉晶的影響研究
市場上銷售的達比加群酯甲磺酸鹽藥物是一種膠囊����,無水晶型Ⅰ在異丙醇和羥丙基纖維素衍生物中分散形成懸浮液中直接噴在酒石酸丸上����。在此過程中����,當原料藥分散在異丙醇中時����,也可能發(fā)生無水晶型I向晶型II的轉化����。如上所述,異丙醇是一種溶劑����,其中達比加群酯甲磺酸至少具有一定的溶解度����。為了更好地理解這一生產步驟中的多晶型轉化,進行了幾項小型實驗室實驗����,模擬了異丙醇懸浮液的制備條件和甲磺酸達比加群酯的成球過程。在這些小規(guī)模的實驗室實驗中����,不同組分(原料藥、異丙醇����、羥丙基纖維素)的比例反映了組分的比例����。這些探索性實驗室規(guī)模實驗的結果總結如下:正如預期的那樣,觀察到強烈的時間����,溫度和初始無水晶型II含量依賴性。一般來說,在較高的溫度和較高的初始晶型II水平下����,轉化率隨著時間的推移而增加。在制備達比加群酯甲磺酸懸浮液前����,將噴霧懸浮液輔料羥丙基纖維素溶解于異丙醇中,轉化率降低����。在異丙醇中懸浮的純I型原料藥批次(小于0.6%的II型原料藥(DSC定量限))保存:A)在30℃下保存48小時;或B)在15℃下可達96 h,所得懸浮液中未檢測到II型(拉曼法LOQ為5%)����。然而����,如果同一批次在30°C下懸浮48小時以上,則觀察到16%的轉變?yōu)镮I型����。需要注意的是,實驗室比例模型模擬的是最壞的情況����,因為整個懸浮液被攪拌而沒有噴射����,從而減少了懸浮液的體積。在大規(guī)模生產中����,攪拌好的懸浮液被連續(xù)噴灑,從而減少了懸浮液的體積����,因此只有最后一部分在高達30°C的懸浮中停留48小時。因此����,預計最終顆粒生產過程中產生的晶型II含量將顯著低于實驗室模型。且藥品穩(wěn)定性研究表明����,在儲存期間沒有從I型轉化為II型����。
2.2.2 原料和制劑成品的穩(wěn)定性放置過程中轉晶風險評價
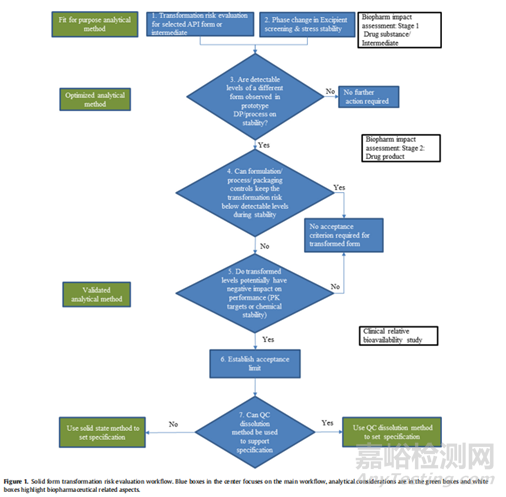
在這一階段����,該評估是在典型穩(wěn)定性留樣條件下進行的����,可以提供對轉化風險和程度的定量了解。期望的結果是根據(jù)已開發(fā)的定量分析方法的檢測限度證明沒有發(fā)現(xiàn)晶型轉化過程����。如果在制劑中檢測到晶型變化,接下來的步驟是進行適當?shù)难芯?���,了解晶型轉化的相關觸發(fā)因素,并重新設計配方和/或工藝和/或存儲條件����,以調節(jié)或消除這些風險因素����。因此,這一步的預期結果是在藥品的生產過程中抑制晶型轉化����。危險因素的例子包括為轉化提供良好微環(huán)境pH值的賦形劑����、生產過程中使用的水分、或引入機械應力的高能單元操作����。這些機制方面的考慮很重要����,因為它們有助于確定晶型的控制戰(zhàn)略����。如果開發(fā)研究導致了配方和/或工藝修改的實施����,并證明在生產或穩(wěn)定過程中沒有可檢測到的晶體形態(tài)轉變,則在藥品中不要求建立晶體形態(tài)的驗收標準����。為了確認修改后的配方和/或工藝和/或存儲條件一致地減輕了晶型轉化的風險����,可能會繼續(xù)監(jiān)測未來的藥品批次。被監(jiān)控的批次應包括規(guī)模擴大����、技術轉移和主要穩(wěn)定性批次����,因為一些與工藝相關的晶體形態(tài)轉化可能取決于規(guī)模的大小。
2.3多晶型的溶解性����、固有溶出速率或溶出度評價
如果溶解方法能夠以適當?shù)撵`敏度檢測到不同晶型形式的變化����,則多晶型的溶解性、固有溶出速率或溶出度研究可作為體外晶型物質評價的輔助方法����。但需要注意一點:溶出度是一個測量藥物從配方基質中體外釋放的過程����。除藥物本身的溶解度以外,溶出還受潤濕性����、粒徑等配方或工藝參數(shù)的影響,因此溶出速率的變化機制是混亂的����,與晶體形態(tài)變化沒有具體聯(lián)系。溶出試驗對晶體變化的敏感性也可能取決于化合物的溶解度����。溶出不太可能區(qū)分高溶解度化合物的不同晶型形式。對于溶解性差的化合物����,如果它們的溶解度存在足夠的差異����,則可以選擇一種溶出介質來區(qū)分它們的不同晶型形式。
開發(fā)一種QC溶出方法����,能夠以可接受的特異性和靈敏度檢測多種晶型形式,往往是一項挑戰(zhàn)����。一般來說����,QC溶出介質需要滿足3倍漏槽條件,以確保在溶出測試結束時藥物可完全釋放。在QC溶出設置中����,通常會減小不同晶型之間的溶解度差異,特別是在溶出介質中使用表面活性劑����,以增強溶解度����,從而可以獲得用于質量控制目的的完全釋放要求����。然而,在某些情況下����,QC溶出方法可以用來監(jiān)測不同晶體形態(tài)的變化����,例如,當在溶出介質中觀察到不同晶型之間的溶解度有顯著差異時����,緩慢或不完全的溶解釋放可能表明存在晶型的轉化行為。開發(fā)可區(qū)分晶型的QC溶出方法時����,文獻中提供以下的工作流程可供參考:
1)在生理pH值范圍(1.0 ~ 7.5)和生物相關的介質中,收集每個純晶型在多種介質中的平衡溶解度數(shù)據(jù)����。如果在溶解度測量過程中發(fā)生了晶體變化����,應該注意的是,溶解度值代表的是轉化后的晶體����,而不是最開始投入的晶體形態(tài)。
2)選擇溶解介質并計算劑量/體積比����,以評估是否能夠滿足漏槽條件(3倍平衡溶解度)����。避免或盡量減少表面活性劑的使用,這樣在溶解過程中,不需要的晶體形式可以保持不溶性����。根據(jù)配方選擇合適的溶出設備和攪拌速度����。
3)通過將非目標的多晶形式(即1 - 10%)添加到目標晶型(即90 - 99%)中,支制備成相應的制劑進行定量評估����。根據(jù)選擇的方法條件進行溶出試驗����,并在最終采樣時間點取樣分析����。計算釋放百分比,并評估釋放程度是否與配方中目標晶型的的溶出水平相當����,以確定該方法對檢測配方中不需要的晶型形式是否足夠敏感。
4)如果QC溶出方法能夠在制劑樣品中檢測出合理數(shù)量的非目標晶型形式����,那么它就可以用于晶型的控制策略之一。如果是這種情況����,則適當設置溶出標準,以確保在產品性能可接受的水平上檢測和控制不良的晶型轉化形式����。否則,仍將需要開發(fā)固態(tài)相關檢測技術����。
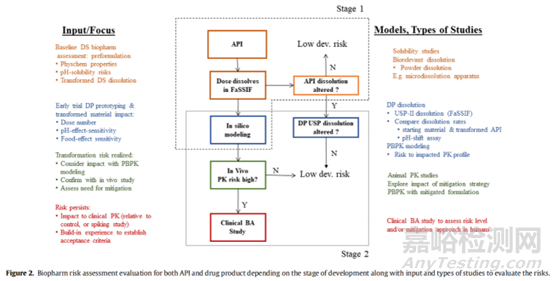
3、多晶型藥物的生物學評價
為了確認晶型轉化風險對體內有效性/安全性造成的影響����,而不是過度區(qū)分體外溶出方法學的產物����,必要時也可以進行動物PK研究。通常����,狗和犬科動物模型是常用的,大鼠和迷你豬也可以考慮����。在轉化風險評估的早期階段,可在大鼠體內研究不同晶型摻雜比例的原料藥混懸劑配方����。對于口服固體劑型的晶型轉化研究,狗和犬科動物在禁食狀態(tài)����、喂食狀態(tài)或pH值改變狀態(tài)下的研究是最具代表性的給藥經驗����。各種模型的原料藥和劑量轉化����、晶體形態(tài)轉化風險和對人體生物性能的預測程度都有已公布藥物的例子,大家可以自行查閱參考����。
臨床研究:臨床相對生物利用度研究對于建立和/或可接受的晶型轉化形式的水平也很有價值����。FDA關于生物利用度和生物等效性的指南提供了關于相對于參考產品進行此類轉化的臨床研究的詳細信息����。通常����,這是一種多交叉的相對生物利用度研究����,將已知(加標)晶型轉化原料藥水平的試驗劑型與低轉化原料藥或無晶型轉化原料藥水平的參考劑型進行比較。有文獻提出了一種體外生物相關溶出����、計算機建模、動物PK和臨床PK評估的生物學評價工作流程是監(jiān)管機構建立和認可的工具����,用于評估患者體內的晶型轉化風險����,并證明降低風險策略的有效性。
例如達比加群酯甲磺酸鹽在兩項臨床研究中評估了晶型II對藥品相對生物利用度的影響����。在一項雙向交叉、隨機����、開放試驗中進行的相對生物利用度研究沒有證明純劑型I和純劑型II批次之間的生物等效性。兩種形式的PK差異很小����,但具有統(tǒng)計學意義,無水晶型II的Cmax和AUC略低����。然而����,第二次生物等效性研究表明����,含有17%晶型II的試驗批次與參考純晶型I批次之間具有生物等效性。因此����,最終藥品中可以接受一定量的晶型II,并根據(jù)此結果制定原料藥和制劑的晶型控制策略����。
參考文獻:
[1] P. Sieger , U. Werthmann , S. Saouane ,The polymorph landscape of dabigatran etexilate mesylate: Taking the challenge to bring a metastable polymorph to market [J]European Journal of Pharmaceutical Sciences ,186 (2023) 106447
[2] A. Bauer-Brandl,Polymorphic transitions of cimetidine during manufacture of solid dosage forms[J]International Journal of Pharmaceutics 140 (1996) 195-206
[3] Umesh Kestura, Anisha Patela, Sherif Badawya, Neil Mathiasa, Limin Zhangb ,Strategies for Managing Solid Form Transformation Risk in Drug Product[J]Journal of Pharmaceutical Sciences 112 (2023) 909−921