摘 要 / Abstract
我國正處于《藥品注冊管理辦法》實施的初始階段�����,制定全面�����、完善的藥品注冊核查體系和機制���,有助于規(guī)范基于風險啟動注冊核查的程序�����,提升我國注冊核查啟動情形制定的科學性和完整性�,推進基于風險的審評�、核查并聯的進程,提高注冊審評審批的工作效率。本文詳細介紹了FDA 和EMA 有關基于風險啟動批準前檢查的情形及風險考慮因素, 并結合我國現階段基于風險啟動藥品注冊核查的工作實際�,提出完善注冊核查啟動工作的建議,以期為進一步完善我國基于風險啟動注冊核查情形的選擇和程序提供參考�。
China is in the initial stage of implementing the new version of drug registration management measures. Developing a comprehensive and complete drug registration inspection system and mechanism helps standardize the risk-based registration inspection process, improve the scientific and complete development of registration inspection initiation status in China, promote the parallel process of risk-based evaluation and verification, and improve the efficiency of registration review and approval. This article provides a detailed introduction to the situation and risk considerations of risk-based pre-approval inspections by the FDA and the EMA. Based on the current situation of risk-based drug registration inspections in China, it offers references for further improving the selection and procedures of risk-based registration inspection in China.
關 鍵 詞 / Key words
藥品;注冊核查�����;基于風險�����;啟動情形�����;FDA �;EMA
drug; registration inspections; risk-based; initiation status; FDA; EMA
我國實施藥品審評審批制度改革以來,國家藥品監(jiān)督管理部門陸續(xù)發(fā)布了一系列審評審批制度改革的配套文件[1-9]�,提出了優(yōu)化審評審批流程,加快藥品審評審批�����;建立以審評為中心�,核查、檢驗為支撐的技術審評體系���,逐步與國際接軌的藥品監(jiān)管科學體系等要求�����。提高藥品審評審批效率���,按時限審評成為常態(tài)要求,其中審評流程由省級食品藥品監(jiān)督管理部門受理�����、原國家食品藥品監(jiān)督管理總局審評審批的藥品注冊申請�,調整為國家藥品監(jiān)督管理局集中受理。集中受理后���,新受理的注冊申請�����,將根據藥品技術審評的需求由國家藥品監(jiān)督管理局食品藥品審核查驗中心統一組織全國藥品注冊檢查資源實施現場核查[10]�。注冊核查模式由“逢審必查”向基于風險啟動轉變���,基于風險啟動藥品注冊核查是進一步優(yōu)化注冊程序的主要節(jié)點�����,是實現審評審批總時限可預期的重要保證[11]�。
基于風險啟動藥品注冊核查是適應藥品高質量發(fā)展的需要,其既能夠控制注冊審評階段對安全性���、有效性�、質量可控性的風險�����,又能夠節(jié)約人力�、物力,提高審評審批效率�����,提升行業(yè)的發(fā)展速度�。因此,為了建立規(guī)范有效的合規(guī)審查體系�,基于風險啟動注冊核查的原則和程序就顯得尤為重要。我國已初步建立了基于風險啟動注冊核查的標準與程序�,但仍在不斷完善和提高,以期與國際接軌�����,建立更加科學���、有效的適合我國國情的注冊核查啟動模式�����。本文介紹了美國食品藥品監(jiān)督管理局(Food and Drug Administration���,FDA)以及歐洲藥品管理局(European Medicines Agency,EMA) 基于風險啟動批準前檢查的情形和風險評估考慮因素�,并提出了相關建議,為完善我國注冊申請���、啟動注冊核查提供參考與思路���。
1、FDA 基于風險啟動注冊核查的概述
2002 年8 月21 日�,FDA 啟動了“21 世紀制藥cGMP :基于風險的方法(Pharmaceutical cGMP for the 21stCentury: A Risk-Based Approach)”倡議,提出鼓勵實施基于風險的方法�����,將行業(yè)和機構的注意力集中到關鍵領域,確保監(jiān)管審查和檢查政策以最先進的制藥科學為基礎[12]���。2022 年9 月FDA 更新了《藥品生產檢查合規(guī)項目手冊》(Compliance Program Guidance Mannual)中的《批準前檢查手冊(7346.832)》(7346.832 Pre-approval Inspections- Investigations Revised)[13] 旨在指導對某項申報是否需要進行現場檢查建立標準�,并制定了基于風險的決策標準�����,來判斷是否需要對某項申請進行現場檢查�����,從而保證檢查資源使用到最有可能危害公眾健康的領域���。
1.1 基于風險的情形選擇原則
2010 年版的《批準前檢查手冊(7346.832)》表明�,FDA 的現場檢查包括批準前檢查�����、批準后監(jiān)督檢查���、常規(guī)監(jiān)督檢查和有因檢查�����。批準前檢查(preapproval Inspection�����,PAI)指FDA 在藥品申請審評過程中�����、藥品批準之前���,對藥品申報中所提交的數據的真實性和準確性的核實,及對申請中列明的任何生產設施的現行藥品生產管理規(guī)范(current Good Manufacture Practices�����,cGMP)狀況通過現場檢查和(或)地區(qū)文件審查的方式對生產企業(yè)進行的評估活動�����。進行PAI 有助于FDA 保證藥品申請中所列生產企業(yè)具有生產藥品的能力�,及所提交數據的準確、完整���。PAI 項目描述了3 種基本的檢查目標���,這些目標都要求綜合信息策略以及仔細的現場檢查評估:①為商業(yè)化生產的準備就緒�����;②與申報資料一致�����;③數據可靠性審計���。
藥品批準前檢查的執(zhí)行可分為2 種類型:優(yōu)先檢查和選擇性檢查。如果一個生產企業(yè)適用于一個或多個以下情況�,則這個企業(yè)符合優(yōu)先檢查標準:某生產企業(yè)第一次被列入FDA 的注冊申請中,即從未被檢查過的企業(yè)或者僅針對非注冊申請藥物被檢查過���;申請人首次提交的注冊申請(涵蓋制劑生產和檢驗)���;已批準藥品的首次仿制藥申請(abbreviated new drug application,ANDA�,涵蓋制劑生產和檢驗);含有一種新分子實體的制劑(不適用于補充申請)���;制劑的含量測驗范圍較窄(如標識量95%~105% 的窄治療指數的藥物)或者需采取滴定法配藥的藥品(不適用于補充申請)���;制劑或原料藥(active pharmaceutical ingredients���,API)的生產工藝發(fā)生顯著的變化或者采取了此生產企業(yè)認證之外的新劑型;API來源具有高風險(如API 來源于動物組織)或者預期用途發(fā)生了顯著變化(如API 之前用于非無菌制劑�,但現計劃用于無菌制劑產品)�;提交了很多個申請,或者某些生產地點�����、工藝�����、產品的變更可能會對目前的設施或工藝的控制狀態(tài)造成重大挑戰(zhàn)���;制劑或API 的申請狀態(tài)分類為“不可接受”或者是因2 年內未經過現場檢查���,狀態(tài)未更新(若為控制實驗室此期限為3 年,若為包材與標簽生產商此期限為4 年)�����,或者是為初始申請,或者是化學���、生產和控制(chemical manufacturing control�,CMC)有重大變更的補充申請的批準前檢查�。
選擇性檢查則是除指定的檢查任務外,其他不屬于優(yōu)先檢查標準的情形�����。FDA 的生產和產品質量處(Division of Manufacturing and Product Quality�,DMPQ) 會在企業(yè)評估系統(establishment evaluation system,EES)中輸入一個待評估的“10 日信”請求�����,并轉發(fā)至地區(qū)辦公室�����,根據地區(qū)辦公室的建議確定是否進行批準前檢查�����。對于FDA 地區(qū)辦公室認為不需進行優(yōu)先檢查的,則將在10 日內把不啟動批準前檢查的原因與建議一起輸入至EES 系統�。對于建議需啟動批準前檢查的,地區(qū)辦公室會使用EES 系統自動生成檢查安排�。
1.2 基于風險模型的風險評估
隨著基于風險的方法不斷成熟與完善,現行的《藥品生產檢查合規(guī)項目手冊》強化了FDA 基于風險的方法�,即利用申請中提供的信息和FDA 已有的關于該設施的信息確定是否需要進行檢查。同時在原有PAI 項目描述的3 種基本檢查目標的基礎上增加了第4 個目標�,即藥品研發(fā)質量承諾,其意思是通過評估支持�、定義、管理和持續(xù)評估其有效性以及在支持藥品質量體系(pharmaceutical quality system�,PQS)持續(xù)改進中的用途來評價藥物開發(fā)計劃�。如果FDA 發(fā)現有足夠的可用信息,則可能不需要PAI���。當提交上市申請時�,FDA 藥品審評和研究中心(Center for Drug Evaluation and Research�,CDER)通過組建整體質量評估(integrated quality assessment,IQA)團隊來執(zhí)行質量評估�,啟動批準前設施評價。IQA 團隊提供與藥品相關的以患者為中心和基于風險的質量建議�,包括對生產、加工、包裝���、貯存及檢驗藥品或原料藥的設施的建議�。在執(zhí)行質量評估時�����,IQA 團隊通過評估以下內容來確定申請中列出的設施的PAI 需求:產品風險和生產工藝與設施風險以及申請中提供信息的準確性和可靠性���。
產品知識和風險評估側重于評估與具體產品使用背景(如治療指數�、患者人群�、臨床獲益)下的產品關鍵質量屬性(critical quality attributes,CQA)相關的風險�����。
生產工藝風險評估側重于評估工藝對產品CQA 的影響���,即關鍵變更參數源是否得到識別和解釋�����,各維度的變更是否通過工藝得到了管理�����,以及工藝性能和產品質量屬性是否可以充分滿足并且有效控制�����。良好的產品和工藝評價意味著從患者的角度對質量至關重要的特征已被識別并轉化為產品的CQA�,并且影響CQA 的物料屬性和工藝參數也能夠識別、表征和控制�。
生產設施風險評估側重于評估生產或設施設備經證明的能力及其與上市申請的相關性,其包括但不限于通過評估設施檢查報告(establishment inspection report���,EIR)和依據�,適用的現場警示報告(field alert report�,FAR), 相關的召回�,監(jiān)管�����、告誡行動以及可用的國外監(jiān)管報告�,審查設施近期的生產歷史。
除以上3個主要風險評估考慮外���,對支持申請的生產場地信息的準確性和可靠性的評估�,也是確定是否需要PAI 的一個重要因素。
另外�����,當需要確認質量數據(其對于確定藥品的安全性���、有效性和質量至關重要)的準確性和可靠性時或經評估需確認設施的操作與注冊申請中申報操作是否相符也可觸發(fā)PAI�,最終也由IQA 團隊根據對申請的綜合風險評估來確定是否需要PAI�����。
1.3 注冊核查啟動流程
CDER 在收到新藥申請(new drug application�����,NDA) 或仿制藥申請(ANDA)的60 日內���,藥品生產評估辦公室(Office of Pharmaceeufacturiong Assesment�,OPMA)向監(jiān)管事務辦公室(Office of Regulatory Affairs���,ORA)發(fā)送PAI 或地區(qū)文件審查(division of filing review�����,DFR) 請求�, 或通過CDER 的信息學平臺軟件Panorama 輸入設施建議。對于PAI 請求:① OPMA通過OPMA 決策或請求任務以明確的理由要求PAI�����,并提供有關已確定的風險和疑慮的檢查策略的特定信息�。② ORA 評估請求,安排檢查并通知OPMA�����。ORA和CDER 應盡可能在應用評估和檢查活動的計劃和時間安排上進行合作���。如果ORA 的評估表明沒有必要進行PAI���,則需與OPMA 共同做出最終決定。在收到請求的10 個工作日內�, ORA 的批準前項目經理(preapproval program manager�����,PAM)需在Panorama 中錄入不啟動檢查的原因以及ORA 的建議。
2�、EMA 基于風險啟動注冊核查的概述
歐盟是由多個成員國組成,藥品的審批程序既要考慮經濟一體化的整體性�����,又要兼顧各成員國獨立自主的個體性�����,因此其藥品審批程序更多樣化���,主要分為成員國審批程序���、集中審批程序、互認可程序���。2001 年���,歐盟對這些指令進行了梳理整合,最終由歐洲議會和歐盟理事會通過了2001/83/EC 號法令《關于人類用藥品的共同體法規(guī)》[14]���,該法令是目前歐盟最全面���、最系統的藥事法規(guī)���。2013 年修訂了附件1 第四部分,引入了“ 基于風險的方法”���。
2.1 基于風險的情形選擇原則
2022 年4 月20 日《歐盟檢查和信息交換程序匯編》(Compilation of Community Procedures on Inspections and Exchange of Information) 進行了修訂[15]�����, 其中《基于風險的藥品生產企業(yè)檢查計劃模型》(A Model for Risk Based Planning for Inspections of Pharmaceutical Manufacturers)提出了通過使用系統的�、基于風險的方法�����,并充分利用其監(jiān)督和執(zhí)法資源�,用于在規(guī)劃藥品生產質量管理規(guī)范(Good Manufacture Practices,GMP) 檢查的頻率和范圍時確定檢查地點的優(yōu)先順序�����?��!痘陲L險的藥品生產企業(yè)檢查計劃模型》適用范圍為成員國主管當局對制劑和原料藥生產企業(yè)的GMP 檢查計劃���,國內和第三國(third country manufacturers) 生產企業(yè)的GMP 檢查計劃,成員國主管當局對研究用藥品生產企業(yè)的GMP 檢查計劃�。但是對于新的生產場地,如果第三國沒有簽署雙邊互認協議( mutual recognition agreements, MRA)并且工廠從未接受過歐盟檢查員檢查�,則將啟動系統地檢查。對于既往已接受過檢查的���,將根據不同生產場地的風險進行評級�,其主要基于2 種類型風險的評估�,即內在風險和合規(guī)風險。
內在風險�,主要是指生產場地的復雜性(規(guī)模和設施種類)、產品制造過程的復雜性(操作類型和順序過程控制)和產品復雜性�����,以及生產場地提供的產品或服務的關鍵性�。值得注意的是,這些項目的復雜性和關鍵性通常保持不變�����。生產場地的復雜性主要包括:生產場地規(guī)模的復雜性�����;現場使用的不同制造或分銷流程的數量;現有設備和設施的配置程度�����;現場工作人員的數量�����;企業(yè)生產提供的商業(yè)市場數量�;企業(yè)生產提供的客戶數量。產品制造過程復雜性主要包括:無菌和無菌制造工藝�����;有高度復雜過程的參數放行程序�;具有大量關鍵步驟的生產工藝;品種劑型為低劑量高溶解性速釋劑���、緩釋劑等制造過程的復雜制劑���;非無菌制造過程中的單元操作數量較大導致過程復雜的;在生產現場進行的后處理或返工的程度。產品復雜性主要包括:包裝組件數量較多的產品���;需要特殊儲存和配送的產品�。
合規(guī)風險���,是基于最近一次GMP 檢查的結果,主要考慮因素為缺陷的分類和數量���,反映了企業(yè)執(zhí)行GMP 的合規(guī)水平�,以及GMP 合規(guī)狀態(tài)等級�。合規(guī)風險的主要考慮因素包括:在最近一次現場檢查中發(fā)現缺陷的區(qū)域,特別是嚴重和主要缺陷�;最近一次現場檢查中未檢查(或未詳細檢查)的區(qū)域;上次檢查時認為資源不足的區(qū)域�����;生產場地的計劃更改可能會改變與生產場地相關的復雜性或關鍵性風險評級���;檢查員認為有必要在下一次檢查的�����。
2.2 基于不同類型風險的評估
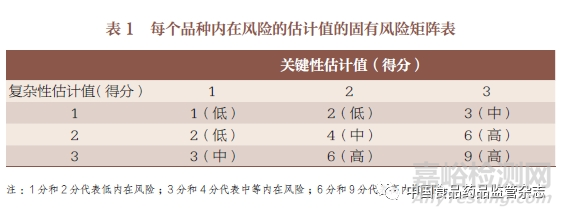
根據內在風險考慮的2 個風險指示因素�,即生產場地、產品制造過程和產品的復雜性以及生產場地提供的產品或服務的關鍵性�。通過每個品種內在風險的估計值的固有風險矩陣可最終判定品種的內在風險等級,見表1���。
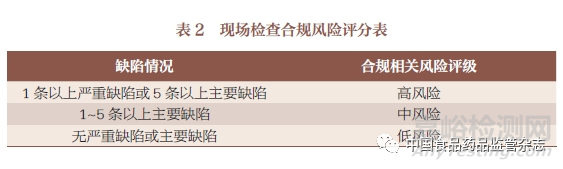
合規(guī)風險的評估是基于最近一次現場檢查(例行或全面檢查���,有因檢查)中與生產場地相關的合規(guī)風險,主要是通過現場檢查中發(fā)現的缺陷���,對其進行梳理分級���,分配相應的高、中或低的等級�,然后再對現場檢查進行合規(guī)風險評級,見表2���。
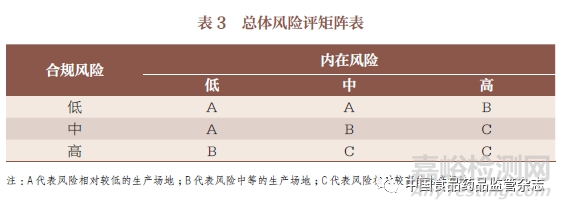
將內在風險和合規(guī)風險利用簡單的矩陣進行組合后生成最終的生產場地的檢查風險等級���,見表3。該結果可用于決定下一次現場檢查的頻次和范圍���。
2.3 基于風險的檢查頻率
根據生產場地的檢查風險等級評級來生成和記錄現場例行檢查的建議頻率���,每個風險評級的建議檢查頻率�����,見表4�����。
風險評級A 的生產場地在固有風險或合規(guī)風險方面至少有一個低風險評分。在例行檢查計劃中�����,這些場所的檢查頻率可能會降低�����,如頻率低于每2 年一次�����。
風險評級為B 的生產場地�,檢查頻率的時間范圍設定為 1~2 年���,但不是絕對的 2 年。例如���,如果2 個生產場地的風險評級為 B�����,但其中一個的最后檢查結果比另一個差(比如存在5 個主要缺陷與一個主要缺陷)�,則分配給前一個生產場地的檢查頻率原則上為檢查頻率規(guī)定的時間范圍的下限(檢查頻率接近1 年而不是2 年)�。
風險評級C 的生產場地在固有風險或合規(guī)風險方面至少有一個高風險評分。在例行檢查計劃中�����,可能會增加檢查這些生產場地的頻率���,至少每年一次或更多的頻率�����。
此外�,分配給具有相同風險評級的生產場地的檢查頻率可能會考慮內在風險和合規(guī)風險各自的風險估計值的分數高低�,從而最終確定該生產場地的檢查頻率�����。例如�����,對于總體風險評級都為C 的生產場地�����,當一個生產場地同時具有高內在風險和高合規(guī)風險時分配的檢查頻率(例如9 個月)可能高于具有高內在風險但中等合規(guī)風險的生產場地�。
上述風險評級目的是保證具有高內在風險評分或高合規(guī)風險評分的生產場地都不會被分配降低的檢查頻率���。分配給任何一個生產場地的實際檢查頻率都能反映上次檢查期間發(fā)現的缺陷的數量和類型。
3�����、我國現場注冊核查啟動程序的現狀
3.1 基于風險的情形選擇原則
《藥品注冊管理辦法》[16] 第四十六條規(guī)定“藥品審評中心根據藥物創(chuàng)新程度�����、藥物研究機構既往接受核查情況等�,基于風險決定是否開展藥品注冊研制現場核查”���,第四十七條規(guī)定“藥品審評中心根據申報注冊的品種、工藝�����、設施���、既往接受核查情況等因素�,基于風險決定是否啟動藥品注冊生產現場核查”���,其配套文件《藥品注冊核查檢驗啟動工作程序(試行)》[17] 基于品種因素和研發(fā)生產主體合規(guī)因素�����,對需納入注冊核查風險等級劃分的情形進行了規(guī)定:①藥品上市許可申請���;②涉及藥品生產過程中處方工藝或生產批量重大變更,或者新增臨床試驗數據等的補充申請�����;③其他需要啟動注冊核查的藥品注冊申請�。
品種因素包括藥物創(chuàng)新程度�����、藥品類型���、工藝和設施等。研發(fā)生產主體合規(guī)因素包括參與藥學研制�����、臨床試驗�、藥理毒理學研究以及生產制造的相關單位和機構既往接受注冊核查與監(jiān)督檢查的情況等。在此基礎上�,將品種和研發(fā)生產主體合規(guī)兩大類評估因素分別劃分為高、中�����、低3種情形�����。根據品種因素和研發(fā)生產主體合規(guī)風險因素具體情形���,經綜合評估研判后�,藥品審評機構將需要注冊核查的藥品注冊申請劃分為高�����、中���、低3 個風險等級�,原則上以品種因素和研發(fā)生產主體合規(guī)因素風險情形較高的確定藥品注冊申請最終風險等級�。
3.2 注冊核查啟動流程
藥品注冊申請原則上在受理后40 日內,由藥品審評機構完成品種因素以及合規(guī)因素的風險標記�����,經綜合評估后確定品種注冊核查風險等級及核查類別(藥學�����、臨床)�����,其中對于高風險等級的藥品注冊申請應當啟動注冊核查�����,對于其他風險等級的藥品注冊申請按比例隨機啟動注冊核查。對于確定啟動注冊核查的藥品注冊申請�,由藥品審評機構準備注冊核查所需相關資料,并在規(guī)定時間內通知核查機構啟動注冊核查�,同時通知申請人,完成啟動注冊核查任務���。
4�、對我國現行藥品注冊核查啟動標準與程序的思考
目前�,我國藥品注冊申請基于風險啟動注冊核查的風險因素考慮方面與發(fā)達國家和地區(qū)的監(jiān)管機構啟動注冊核查的考慮因素類似,均為通過品種因素(藥物創(chuàng)新程度�、品種、工藝�、設施等)和合規(guī)因素(研發(fā)生產主體既往接受檢查情況)兩方面的風險考慮因素,經綜合評估后研判注冊申請的最終核查風險等級�����。誠然�����,我國的藥品注冊核查啟動工作程序已經在多個方面與國際先進水平接軌���,但在進一步提升認知風險���、識別風險的能力,分層次�����、有步驟地進行風險識別、分析和調整等方面仍需完善與提高。探索建立符合我國國情的基于風險啟動核查的合規(guī)風險評估模型���,不斷完善符合我國國情的藥品注冊核查啟動標準與程序,構建藥品注冊核查啟動前基于風險評估的合規(guī)審查體系,才能充分適應不斷發(fā)展�����、不斷進步的藥品監(jiān)管理念�����,提高科學監(jiān)管能力�����,保障公眾用藥安全���。
引用本文
藺娟,喬利濤,周剛*.FDA和EMA基于風險啟動藥品檢查情形概述及對我國的啟示[J].中國食品藥品監(jiān)管,2023(11):60-67.