對于植入和III類器械的制造商申請CE認證來說�����,困擾的首要問題通常都是到底是否需要進行臨床試驗�����;若是不做臨床試驗�����,走等同路徑到底是否一定需要合同?MDCG協(xié)調小組似乎洞察到了大家的這一疑問,這次發(fā)布的指南文件MDCG2023-7直指這兩大敏感問題����。本指南旨在澄清將投放歐洲市場的植入式和III類醫(yī)療器械的臨床試驗要求豁免以及與等效性證明相關的相關條件。它還提供了與根據(jù)MDR附錄XIV第3節(jié)證明“足夠程度的數(shù)據(jù)訪問”相關的例子和考慮因素�����。
臨床試驗要求豁免
對于III類和植入醫(yī)療器械來說����,MDR原文中提到的4種可豁免進行臨床試驗的情況:
1.Article 61(4) 前三個短劃線所指明的情況:
— 該器械由同一制造商對已投放市場的器械進行改造而成;
— 依據(jù)附錄XIV第3節(jié)規(guī)定��,已改進的器械經(jīng)制造商證明后等同于投放市場的器械����,且此證明已得到公告機構認可;
— 對投放市場的器械進行臨床評價足以證明已改進的器械符合相關的安全性能要求
在這種情況下�����,公告機構應核查 PMCF(上市后臨床跟蹤) 計劃是合適的�����,且其中包括上市后研究,以證明器械的安全和能��。此外��,MDR第61條第 6 段所述的情況無需進行臨床研究����。
2.Article 61(6)(a):
此類器械依據(jù)第 90/385/EEC 號指令或第 93/42/EEC 號指令已合法投放市場或投入使用,其臨床評價:
— 基于足夠的臨床數(shù)據(jù)��;
— 符合此類器械臨床評價相關的產品CS(如果有)�����。
3.Article 61(6)(b):
針對縫線��、訂書釘�����、牙齒填充物��、牙套�����、齒冠、螺釘�����、楔子����、板�����、電線��、別針����、小夾和連接器而進行的臨床評價均應建立在充分的臨床數(shù)據(jù)上且符合相關的產品特定的 CS(如果有)。
4.Article 61(5):
依據(jù)MDR第61條第4段規(guī)定��,制造商生產出的器械經(jīng)證實等同于已投放市場的器械(不屬于同一制造商生產)����,除此條所要求的內容外,若以下條件均滿足����,則無需進行臨床研究:
— 在這兩個制造商擬定合同的適當位置明確允許第二種器械制造商在現(xiàn)有基礎上全權使用技術文件����。
— 原始臨床評價已依照MDR要求執(zhí)行����,且第二種器械制造商向公告機構提供其明確證據(jù)。
總結:
對于植入和III類器械來說����,若能滿足如下其中一條,則有可能豁免進行臨床試驗:
● 該產品是本公司已經(jīng)上市產品改型而成�����,且能證明等同�����;
● 該產品已獲取MDD下的CE認證證書����,且基于較為充足的臨床數(shù)據(jù);
● 該產品為WET清單中的器械;
● 與任何一家可以作為等同的器械的制造商簽訂了授權合同��;
獲取數(shù)據(jù)的充分程度
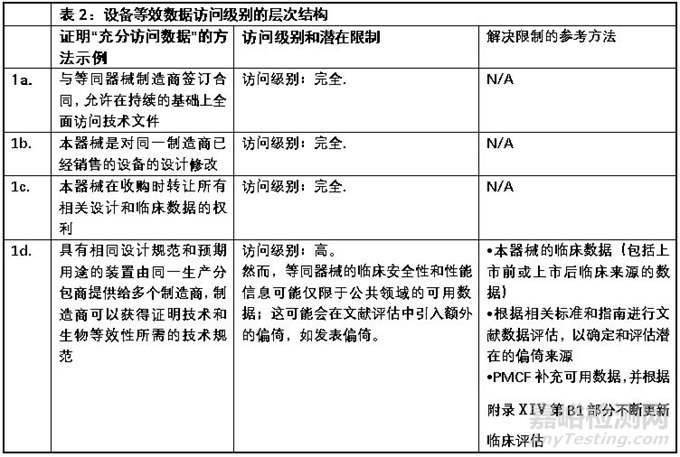
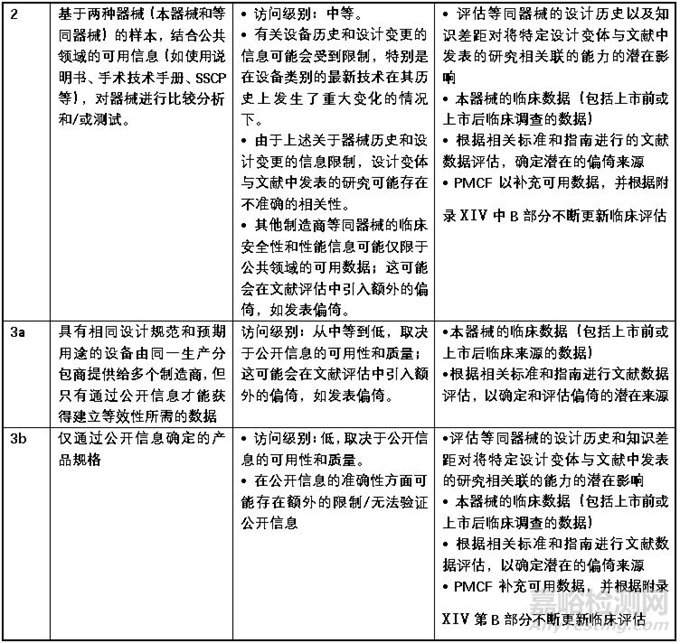
MDCG2023-7強調�����,證明“足夠的訪問水平”并不需要在所有情況下簽訂合同��。合同僅適用于MDR中第61(5)條所述的豁免情況��。還應注意的是��,MDR的附錄XIV第3節(jié)專門提到了證明等效性聲明所需的數(shù)據(jù):即�����,要求有足夠的途徑來確定評估等效性所依據(jù)的臨床�����、技術和生物特征����,而不是獲得完整的技術文件�����。
MDCG2023-7用附表2的形式對“充分訪問數(shù)據(jù)”的方法進行了闡述,該表格內容對公告機構和制造商來說都是評估等同數(shù)據(jù)是否可認為充分獲得的重要參考����。