摘要:目的 探討口服固體制劑工藝驗證中需要關(guān)注的問題,提出建議���,以期為相關(guān)藥品注冊申請?zhí)峁﹨⒖肌?nbsp;
方法 匯總國內(nèi)外藥品監(jiān)管機構(gòu)發(fā)布的法規(guī)和指南中關(guān)于工藝驗證文件的一般要求��,進一步結(jié)合藥品審評實際���,分析口服固體 制劑注冊申報資料中工藝驗證常見問題。
結(jié)果與結(jié)論 工藝驗證是藥品生產(chǎn)質(zhì)量管理規(guī)范( GMP) 一項重要工作,也是支撐藥品上市申請的關(guān)鍵性技術(shù)資料。 藥品生產(chǎn)企業(yè)和研發(fā)機構(gòu)應(yīng)結(jié)合產(chǎn)品具體情況及工藝特點進行詳細研究�,提升工藝驗證質(zhì)量���。
中國藥品生產(chǎn)質(zhì)量管理規(guī)范( GMP) 規(guī)定,工藝驗證應(yīng)當(dāng)證明一個生產(chǎn)工藝按照規(guī)定的工藝參數(shù)能 夠持續(xù)生產(chǎn)出符合預(yù)定用途和注冊要求的產(chǎn)品�。 工 藝驗證應(yīng)當(dāng)包括首次驗證、影響產(chǎn)品質(zhì)量的重大變更后的驗證�����、必要的再驗證以及在產(chǎn)品生命周期中的持續(xù)工藝確認(rèn)�,以確保工藝始終處于驗證狀 態(tài)[1] ��。 美國食品藥品監(jiān)督管理局( FDA) [2] 、世界衛(wèi)生組織(WHO)[3] �、歐洲藥品管理局(EMA)[4?5] 將工藝驗證劃分為 3 個階段,包括第一階段工藝設(shè)計 ( process design) ���、第二階段工藝驗證( process qualifi? cation/ process validation)�����、第三階段持續(xù)工藝確認(rèn) ( continued process verification / on?going process verification) ���。 可見工藝驗證貫穿于整個產(chǎn)品生命周期已成為國際上的共識,工藝驗證對于保證藥品質(zhì)量可 控�、安全、有效具有重要意義�。 2020年我國新修訂的《藥品注冊管理辦法》[6] 要求申請人完成商業(yè)化生產(chǎn)規(guī)模生產(chǎn)工藝驗證后才可以提出藥品上市許可申請。
筆者匯總了各國藥品監(jiān)管機構(gòu)關(guān)于工藝驗證的相關(guān)要求��,并結(jié)合審評經(jīng)驗��,對口服固體制劑藥品上市申請?zhí)峤坏墓に囼炞C資料中常見的問題進行探 討��,以期為藥品注冊中工藝驗證資料的整理提供參考����。
1���、 工藝驗證文件的基本要求
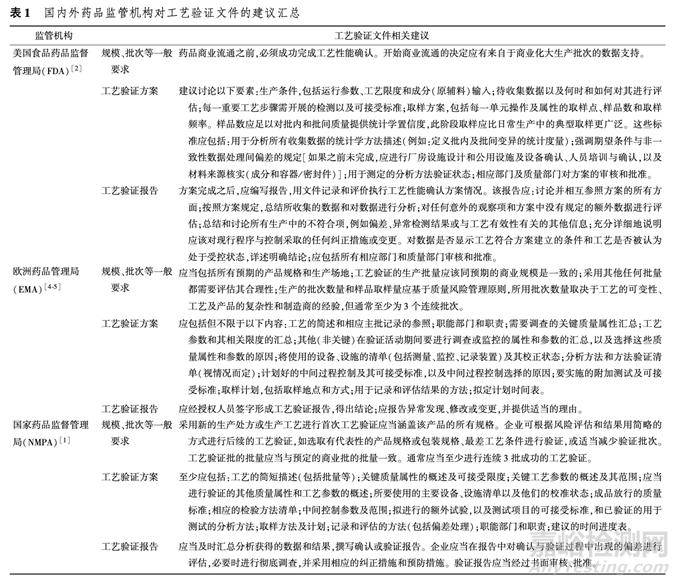
工藝驗證是一項持續(xù)性的工作,需要收集并評估從產(chǎn)品研發(fā)���、商業(yè)化生產(chǎn)工藝確認(rèn)��,到獲準(zhǔn)上市后日常商業(yè)化生產(chǎn)中的持續(xù)確認(rèn)的數(shù)據(jù)�����,使工藝處于 受控狀態(tài)�����。 工藝驗證是 GMP 的重要組成部分�����,國家藥品監(jiān)督管理局( NMPA) 食品藥品審核查驗中心在國內(nèi)外進行藥品生產(chǎn)現(xiàn)場檢查時���,發(fā)現(xiàn)在工藝驗證方面存在較多缺陷[7] 。 就藥品上市申請來講���,注冊申報資料中提供的工藝驗證相關(guān)文件較為有限�,主要是針對新產(chǎn)品的工藝性能確認(rèn)��,筆者將對此進行重點分析����。 對于用于工藝驗證的設(shè)備、設(shè)施����、公用系統(tǒng)的確認(rèn)等一般在現(xiàn)場檢查時予以關(guān)注;對于產(chǎn)品上 市后的變更工藝確認(rèn)、持續(xù)工藝確認(rèn)則為藥品上市后監(jiān)管的內(nèi)容;上述兩方面不作為本文討論的重點�����。國內(nèi)外相關(guān)法規(guī)和指南對工藝驗證文件的要求類似��,見表 1�����。
EMA 和 NMPA 對工藝驗證的批次和批量提出了要求��,而 FDA 則沒有明確規(guī)定�����。 國內(nèi)外藥品監(jiān)管機構(gòu)頒布的法規(guī)和指南是對工藝驗證的一般要求和 相關(guān)文件撰寫的概括性建議。 在實際生產(chǎn)和運用中�����,可按上述要求搭建工藝驗證框架���,但仍需根據(jù)具體品種特點和工藝特性制定更為具體和可行的驗證 計劃���。
2、 口服固體制劑注冊申報工藝驗證常見問題考量
目前藥品注冊資料中����,申請人一般連續(xù)生產(chǎn)3批次驗證批產(chǎn)品,對工藝參數(shù)��、中間體和成品質(zhì)量檢測�、收率和物料平衡等數(shù)據(jù)匯總,形成工藝驗證文件進行提交�����,用以支持產(chǎn)品的上市申請���。 但工藝驗證過程中通常需要比在常規(guī)生產(chǎn)中進行更多的取樣和監(jiān)控���,以更好地確認(rèn)工藝性能及確證產(chǎn)品質(zhì)量的一 致性���,從而建立常規(guī)取樣和對特定產(chǎn)品及工藝的監(jiān)測水平和頻率�。 以下就口服固體制劑注冊申報中提供的工藝驗證資料常見問題進行分析,并提出建議����。
2. 1 制劑生產(chǎn)工藝存在亞批
部分產(chǎn)品由于市場需求擬定較大的商業(yè)化生產(chǎn)批量,但受車間���、設(shè)備生產(chǎn)能力所限���,可能采用分亞 批生產(chǎn)的形式。 驗證亞批之間的質(zhì)量均一性對于保 證終產(chǎn)品質(zhì)量具有重要影響����,也是目前注冊申報中 容易忽略的問題,審評對于該類問題具有較高的發(fā)補率�。
例如某口服固體制劑采用濕法制粒生產(chǎn)工藝,按照批處方將制粒所用物料均分為 4 等份���,分 4 鍋分別進行預(yù)混��、制粒���、干燥���、整粒工序生產(chǎn),然后將 4 鍋干顆?��;旌?����,再與外加崩解劑�、潤滑劑等總混��,壓片�,包衣。 盡管每一鍋采用相同的工藝參數(shù)�,但為證明工藝穩(wěn)定性并保證終產(chǎn)品的質(zhì)量均 一,建議在制定工藝驗證方案取樣計劃時����,對制粒的每一亞批物料均取樣檢測顆粒的水分、含量,以及顆粒粉體學(xué)性質(zhì)如粒度分布�����、堆密度���、振實密度 等��。 對收集到的數(shù)據(jù)進行分析和評價,如計算相對標(biāo)準(zhǔn)偏差(RSD)或絕對差值等��,驗證各亞批顆粒之間的質(zhì)量均一性�����,以減少質(zhì)量風(fēng)險���,為擬定商 業(yè)化生產(chǎn)規(guī)模提供依據(jù)��。
2. 2 含量均勻度風(fēng)險
某些產(chǎn)品經(jīng)風(fēng)險評估混合或壓片����、填充工藝步驟為中高風(fēng)險���,應(yīng)在工藝驗證中對混合均勻度和中控劑量單位均勻度進行深入研究��,以確保終產(chǎn)品的含量均勻性�����。 國內(nèi)制藥企業(yè)在進行工藝 驗證中���,一般都進行了總混混合均勻性研究�����,但 容易忽略預(yù)混混合均勻性研究�����,對于某些采用粉末混合直接壓片工藝或規(guī)格相對較小的制劑��,含量均勻性風(fēng)險較大���,建議進行預(yù)混混合均勻性研 究。 在壓片或填充階段以片重�����、片重差異/裝量 差異等考察壓片或填充工藝的穩(wěn)定性,未進行劑量單位均勻度考察���。
參照《化藥口服固體制劑混合均勻度和中控劑量單位均勻度研究技術(shù)指導(dǎo)原則》[8] �����,建議在混合階段制定取樣計劃時���,取樣點應(yīng)分布均勻且具有代表性,至少選取 10 個取樣點�����,每個取樣點至少取 3 份樣品��。 壓片 / 填充工序取樣點應(yīng)覆蓋整個壓片 / 填充過程�����,建議整個批次中一般不少于 20 個取樣點�����, 每個取樣點至少取樣 7 個劑量單位�����。 對所有取樣點的單值���、平均值以及 RSD 進行測定分析��。
此外���,部分企業(yè)的工藝可能存在卸分料的過程, 物料轉(zhuǎn)移至壓片或填充工序前�����,需要分卸到中轉(zhuǎn)容 器中���。 由于物料顆粒大小��、密度的不同�����,可能會導(dǎo)致 物料分層��,因此也建議對卸料過程進行驗證�����,根據(jù)中轉(zhuǎn)容器的構(gòu)造��,設(shè)計具有代表性的取樣點��,驗收標(biāo)準(zhǔn)可參考上述指導(dǎo)原則�����。
2. 3 復(fù)雜工藝制劑
大部分口服固體制劑生產(chǎn)工藝相對較為簡單��,
生產(chǎn)企業(yè)對工藝研發(fā)和工藝驗證也具有較多的經(jīng)驗�����。 但也有一些復(fù)雜工藝的制劑�����,在工藝驗證方面 容易存在缺陷��。
例如某緩釋片劑��,是采用活性成分的膜控包衣緩釋微丸�����,與外加輔料混合壓片制成�����,每個微丸是一個獨立控制的遞藥單元�����。 緩釋微丸的制備過 程中��,可能采用空白丸芯上藥���、干法制粒��、濕法制 ?����;驍D出滾圓工藝制備含藥小丸��,然后膜控包衣���。 無論采用何種工藝���,含藥小丸和緩釋微丸的粒度 分布都應(yīng)在工藝驗證中重點關(guān)注,其對于微丸之間的均一性�����、后續(xù)與外加輔料的混合均勻性乃至 終產(chǎn)品的含量均勻性都具有較大影響��。 建議對各 步驟中間體收集到的粒度分布數(shù)據(jù)進行充分的分析���,以加深對產(chǎn)品工藝的理解�����,為商業(yè)化生產(chǎn)的中 控指標(biāo)提供依據(jù)��。
此外�����,該制劑另一難點是要保證壓片前后溶出行為一致��,確保壓片工藝不會造成緩釋微丸包衣層 結(jié)構(gòu)的破壞。 除處方因素外���,壓片過程中壓力控制 較為關(guān)鍵�����,一般在工藝開發(fā)實驗室或中試規(guī)模下考 察不同硬度范圍內(nèi)的溶出度或溶出曲線��,從而確定壓片工藝的壓力��、片劑硬度等���,在工藝驗證中需對商 業(yè)化規(guī)模的壓片參數(shù)進行進一步確認(rèn)���。 由于其工藝的特殊性,建議對擬定的壓力范圍進行驗證�����,同時應(yīng) 較擬定商業(yè)化生產(chǎn)增加壓片過程檢測的取樣點��,對 片劑的溶出行為進行考察分析���,以驗證壓片工藝的 穩(wěn)定性���,減少制劑溶出質(zhì)量風(fēng)險���。
2. 4 中間產(chǎn)品貯存時限
目前部分藥品注冊申請中,未能提供中間產(chǎn)品貯存時限的相關(guān)的研究資料�����,或提供的驗證資料不全面�����,如未對黏合劑溶液或者包衣溶液的貯存時限 進行考察��,總混顆粒貯存時限研究缺少粉體性質(zhì)考察��、微生物限度考察等�����。
國內(nèi)外 GMP 均提及中間產(chǎn)品和待包裝產(chǎn)品應(yīng)當(dāng)在適當(dāng)?shù)臈l件下貯存�����,應(yīng)對生產(chǎn)的每一階段設(shè)定完成時限��,以確保藥品質(zhì)量。 制藥企業(yè)在制 劑生產(chǎn)過程中�����,不可避免地存在中間產(chǎn)品短暫貯 存的情況�����,應(yīng)該確定中間產(chǎn)品可接受的保存期 限��,以確保在下一工藝步驟前符合相應(yīng)的質(zhì)量標(biāo)準(zhǔn)�����。 參照 WHO 保持時間研究一般指南[9] �����,中間產(chǎn)品的貯存時限通常應(yīng)該在產(chǎn)品上市前確定���,可在產(chǎn)品開發(fā)中試規(guī)模或批量放大過程中進行�����,并 在商業(yè)化生產(chǎn)規(guī)模工藝驗證中進行確認(rèn)。 可采用一批至多批樣品�����,或根據(jù)風(fēng)險評估確定研究所 用的批次數(shù)量��。 應(yīng)采用與實際生產(chǎn)階段相一致的環(huán)境條件和容器���,或采用相同材質(zhì)的模擬容器��。 建議對獲得的數(shù)據(jù)進行統(tǒng)計學(xué)分析��,以確定趨勢���,并證明貯存時限設(shè)置的合理性。 以包衣片為例���,提供了可考慮進行貯存時限研究的中間產(chǎn) 品及考察指標(biāo)作為參考���,見表 2。
2. 5 多規(guī)格產(chǎn)品工藝驗證
部分產(chǎn)品同時申報多個規(guī)格�����,存在中間體共用的情況,容易出現(xiàn)驗證批次不夠�����,驗證批量與擬定商業(yè)化生產(chǎn)批量不一致的問題��。 例如某片劑兩個規(guī)格 產(chǎn)品處方比例一致�����,制粒工藝一致��,將一批顆粒與外加物料總混后分為兩份�����,分別按照不同的劑量和壓片參數(shù)進行兩種規(guī)格的壓片�����。 應(yīng)對中間體的生產(chǎn)過程進行連續(xù) 3 批生產(chǎn)工藝驗證��,并且對每一申報規(guī)格分別進行 3 批次壓片工藝驗證��,每一規(guī)格壓片的批量應(yīng)與擬定的商業(yè)化生產(chǎn)規(guī)模一致���,并符合注冊批量的要求��。 若商業(yè)化生產(chǎn)可能存在采用一批總混物料僅進行 1 個規(guī)格壓片的情況��,則在申報資料中同樣需要提供該情況下的工藝驗證資料���,以證明壓片工藝的穩(wěn)定性。
2. 6 原料藥粉碎工藝驗證
原料藥粒度分布是口服固體制劑產(chǎn)品開發(fā)中需要重點關(guān)注的內(nèi)容之一�����,如原料藥粒度不同流動性存在差異�����,可能會影響制劑產(chǎn)品含量均勻性; 原料藥溶解性差�����,粒度分布可能會影響制劑溶出特性;最終可能會影響藥品質(zhì)量�����、安全性和有效性�����。 若制劑生產(chǎn)工藝對原料藥粒度分布有要求, 則需要對原料藥的粉碎工藝進行驗證��,目前部分申報資料僅僅是對粉碎后的原料藥進行粒度分布檢測�����,符合要求即為通過驗證���,實際上并不能足夠保證粉碎工藝的穩(wěn)定性。 建議根據(jù)采用的設(shè)備原 理制定相應(yīng)的粉碎工藝參數(shù)��,如氣流粉碎機物料加料速度�����、氣流壓力;萬能粉碎機粉碎轉(zhuǎn)速和篩網(wǎng)孔徑;在商業(yè)化生產(chǎn)規(guī)模下對工藝參數(shù)范圍進行驗證��,關(guān)注粉碎前后篩網(wǎng)完整性��,粉碎工藝結(jié)束后 多點取樣測定粒度分布�����,工藝參數(shù)控制與粒度檢 測相結(jié)合確保原料藥粒度分布達到預(yù)定要求。 另外��,由于物料放置過程中可能會結(jié)塊或聚集��,改變 已粉碎物料的粒度分布���,因此建議考察粉碎后物料的貯存期�����。
2. 7 異常情況和偏差
注冊申報資料中提交的工藝驗證資料往往是驗證成功的批次��,較少有異常情況統(tǒng)計和偏差記錄���。 部分存在異常和偏差,但工藝驗證報告中僅對偏差的發(fā)生和調(diào)查結(jié)果進行簡單歸納�����,未能提供詳細的偏差調(diào)查資料�����,或偏差調(diào)查不徹底���,不能完全解釋偏差發(fā)生的原因��。
實際上工藝驗證中可能存在放大效應(yīng)�����、工藝操作不熟練��、環(huán)境變化等��,驗證發(fā)生偏差是可以理 解的���,在此階段進行嚴(yán)謹(jǐn)深入的調(diào)查分析���,找出根 本原因�����,及時記錄和評估�����,制定有效的糾偏和預(yù)防措施���,作為工藝驗證報告的附件呈現(xiàn)在注冊申報資料中���,可為后續(xù)產(chǎn)品上市和商業(yè)化生產(chǎn)提供足夠的支持��。 另外工藝驗證階段發(fā)生異常并不可怕�����,反而是工藝參數(shù)范圍的進一步調(diào)整優(yōu)化���,例如某片劑在工藝驗證時,發(fā)現(xiàn)片劑脆碎度不符合規(guī)定��,分析原因為在擬定的壓片機主壓壓力下限壓片導(dǎo)致��,據(jù)此將商業(yè)化生產(chǎn)壓片工藝的主壓壓力 收緊���,以減少質(zhì)量風(fēng)險�����。
2.8 其他
注冊申報工藝驗證資料中�����,以下問題也需要關(guān)注:同一產(chǎn)品擬采用高密度聚乙烯瓶裝和鋁塑泡罩兩種包裝形式���,擬定商業(yè)化生產(chǎn)中采用多臺壓片設(shè) 備���,原輔包具有多個來源等,應(yīng)當(dāng)對每一種情形均進行驗證�����,一并在注冊申報中提供相關(guān)的工藝驗證資料�����。
3��、 結(jié) 語
隨著 GMP 水平的持續(xù)發(fā)展�����,“質(zhì)量源于設(shè)計”“質(zhì)量風(fēng)險管理”等概念越來越多在制藥行業(yè)應(yīng)用��, 只有對生產(chǎn)過程和生產(chǎn)過程的控制進行仔細的設(shè)計 和驗證��,才能夠保證持續(xù)不斷的生產(chǎn)出合格的藥品���。 各國監(jiān)管機構(gòu)關(guān)于工藝驗證的指南為概括性文件��, 提供了一般原則和方法���,而非具體的操作性指南,故對于藥品研發(fā)和生產(chǎn)機構(gòu)來講��,需要結(jié)合具體產(chǎn)品 特性和對其工藝的理解程度來制定可行的工藝驗證方案并進行實施��,以獲得足夠多的數(shù)據(jù)對生產(chǎn)工藝 和產(chǎn)品質(zhì)量進行評價和分析��,確保藥品的質(zhì)量可控���、 安全和有效�����。方案并進行實施�����,以獲得足夠多的數(shù)據(jù)對生產(chǎn)工藝 和產(chǎn)品質(zhì)量進行評價和分析�����,確保藥品的質(zhì)量可控��、 安全和有效�����。