無菌藥品作為高風險產(chǎn)品�,生產(chǎn)過程和質(zhì)量控制具有復雜性和特殊性����,歐盟《人用和獸用藥品生產(chǎn)質(zhì)量管理規(guī)范指南》附錄1“無菌藥品生產(chǎn)”(歐盟GMP附錄1) 的發(fā)布對全球無菌藥品的質(zhì)量管理產(chǎn)生了巨大影響。為了解國際無菌藥品生產(chǎn)質(zhì)量管理的最新發(fā)展趨勢與要求���,把握我國無菌藥品質(zhì)量管理的現(xiàn)狀��,促進我國無菌藥品質(zhì)量提升����,本文對歐盟與我國《藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂)》附錄1“無菌藥品”(中國GMP附錄1) 的主要差異點進行了分析和探討����,為我國GMP 附錄1的修訂和無菌藥品生產(chǎn)企業(yè)質(zhì)量提升提供參考。
歐盟《人用和獸用藥品生產(chǎn)質(zhì)量管理規(guī)范指南》附錄1“無菌藥品生產(chǎn)”(以下簡稱“歐盟GMP附錄1”) 于2022年8月25日正式發(fā)布,這是歐盟2015年發(fā)布對無菌藥品附錄修訂的概念文件草案后歷經(jīng)7年最終修訂完成的[1]�����。該附錄要求的最后實施期限是2023年8月25日���,需要指出的是�,附錄中的第8.123條“凍干機和相關(guān)產(chǎn)品轉(zhuǎn)移和裝載/卸載區(qū)域應經(jīng)過設計,盡可能減少操作人員的干預”的最后實施期限為2024年8月25日�。歐盟GMP附錄1首次頒布是在1971年,之后經(jīng)過多次修訂�,上一次修訂是2008年,該版本是與我國現(xiàn)行《藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂) 》的附錄1“無菌藥品”(以下簡稱“中國GMP附錄1”) 內(nèi)容較為接近的版本[2-3]����。
本次歐盟GMP附錄1修訂版從覆蓋范圍�、章節(jié)以及內(nèi)容都有非常大的調(diào)整,為首次全面修訂���。該附錄規(guī)定了無菌藥品生產(chǎn)質(zhì)量控制的基本原則,詳細說明了對無菌藥品生產(chǎn)企業(yè)質(zhì)量體系的要求���,應“確保所有活動得到有效控制�,以盡可能減少無菌產(chǎn)品中的微生物�、微粒和內(nèi)毒素/熱原污染風險”,明確了潔凈廠房����、設備、人員�����、生產(chǎn)等方面的具體要求[4]。我國GMP附錄1自2010年至今已有13年未修訂�,難以滿足當今無菌藥品GMP管理的要求。
2022年版歐盟GMP附錄1是由世界衛(wèi)生組織(WHO) �、歐洲藥品管理(EMA) 和國際藥品檢查合作組織(PIC/S) 聯(lián)合起草,有利于全球標準的協(xié)調(diào)一致�,也將對我國制藥工業(yè)的發(fā)展產(chǎn)生積極而又深遠的影響。學習����、研究和有效轉(zhuǎn)化應用這一附錄,將對我國制藥工業(yè)創(chuàng)新發(fā)展和建立國際化競爭優(yōu)勢發(fā)揮重要促進作用��。
為了解國際無菌藥品生產(chǎn)質(zhì)量管理的最新發(fā)展趨勢與要求���,把握我國無菌藥品質(zhì)量管理的現(xiàn)狀��,促進我國無菌藥品質(zhì)量提升�,本文對比分析了歐盟與我國GMP附錄1的主要差異點�,以期為我國GMP附錄1的修訂和我國無菌藥品生產(chǎn)企業(yè)質(zhì)量提升提供參考。
1�、整體性差異
2022年版歐盟GMP附錄1共11章�, 295條�����,包括范圍�、原則�����、藥品質(zhì)量體系��、廠房����、設備、公用設施���、人員、生產(chǎn)和具體技術(shù)�����、環(huán)境監(jiān)測和工藝監(jiān)測����、質(zhì)量控制�、術(shù)語等[5-6]�。中國GMP附錄1共15章,81條����,包括范圍、原則���、潔凈度級別及監(jiān)測�、隔離操作技術(shù)��、吹灌封技術(shù)�、人員、廠房����、設備、消毒���、生產(chǎn)管理����、滅菌工藝��、滅菌方法、無菌藥品的最終處理�����、質(zhì)量控制����、術(shù)語等。
2008年版歐盟GMP附錄1僅有17頁126條�����,而2022年版則長達59頁����。2022年版本引入了范圍、藥品質(zhì)量體系(pharmaceutical quality system�,PQS) 、公用設施�、環(huán)境和過程監(jiān)測�、術(shù)語表等新的章節(jié),同時加強了質(zhì)量風險管理�、污染控制策略、新技術(shù)和新工藝等結(jié)構(gòu)化內(nèi)容的指導�����,在內(nèi)容上有了大幅增加,各項規(guī)定更加翔實和具體���。2022年版文件還包含一些中國GMP附錄1沒有規(guī)定的內(nèi)容����,如:污染控制策略��、藥品質(zhì)量體系�、公用設施、生產(chǎn)和具體技術(shù)(成型-灌裝-密封技術(shù)�、凍干、密閉系統(tǒng)����、一次性系統(tǒng)) 等。各版本無菌藥品附錄整體性差異對比詳見表1��。
表1 歐盟與我國GMP無菌藥品附錄整體性差異對比
2�、差異分析
歐盟與中國GMP附錄1最主要的差異集中在歐盟附錄1的拓展了適用范圍,明確了新的概念(如質(zhì)量風險管理��、污染控制策略�、藥品質(zhì)量體系等) ���,強調(diào)了新的技術(shù)(如屏障技術(shù)) ,提高了部分技術(shù)標準(如氣流流型測試��、過濾器完整性測試等)�,本部分對兩者的主要差異點進行分析。
2.1 無菌藥品與無菌產(chǎn)品
2022年版歐盟GMP附錄1在范圍章節(jié)中規(guī)定“無菌產(chǎn)品的生產(chǎn)涵蓋多種無菌產(chǎn)品類型(原料藥���,輔料��,內(nèi)包裝材料和成品制劑) ��,包裝規(guī)格(從單劑量到多劑量〉��,工藝(從高度自動化系統(tǒng)到手動工藝) 和技術(shù)(如生物技術(shù)��,傳統(tǒng)小分子生產(chǎn)系統(tǒng)和密閉系統(tǒng)) ”��。中國GMP附錄1中規(guī)定“無菌藥品是指法定藥品標準中列有無菌檢查項目的制劑和原料藥�,包括無菌制劑和無菌原料藥”�����。歐盟GMP附錄1在適用范圍方面從無菌藥品拓展到無菌產(chǎn)品��,除適用于藥品外還適用于輔料�、內(nèi)包裝材料等產(chǎn)品。
2.2 質(zhì)量風險管理(QRM) 和污染控制策略(CCS)
2022年版歐盟GMP附錄1范圍和通則章節(jié)中提出了無菌產(chǎn)品生產(chǎn)中的質(zhì)量風險管理(quality risk management�����,QRM)和污染控制策略(contamination control strategy�����,CCS) 這兩個在中國GMP 附錄1中沒有涉及的概念���。QRM的原則在第一章“范圍”和第二章“原則”中均有描述�����。QRM作為一種識別�、科學評估及控制質(zhì)量潛在風險的前瞻性方法���,應貫穿無菌藥品生產(chǎn)全過程��,包括設施�、設備、系統(tǒng)和規(guī)程的設計和控制的過程��。CCS是指針對微生物�����、內(nèi)毒素/熱原和微粒的一系列有計劃的控制措施���,源于現(xiàn)有產(chǎn)品和工藝的理解并確保工藝性能和產(chǎn)品質(zhì)量���。CCS的控制措施可包括: 原料藥和制劑的物料和組分相關(guān)的參數(shù)和屬性、廠房設施設備的操作條件�����、中間過程控制���、成品質(zhì)量標準以及相關(guān)方法和監(jiān)控頻次等��。CCS是本次附錄修訂新提出的概念���,在第二章“原則”中進行了重點闡述,全文出現(xiàn)多次�,明確需要在整個生產(chǎn)過程和所有關(guān)鍵控制點建立這一概念的原則���。從現(xiàn)狀來看,藥品生產(chǎn)企業(yè)制訂的文件中已經(jīng)包含了許多CCS要素�,但沒有形成系統(tǒng)化的文件��,歐盟GMP 附錄1 要求CCS應為一個多元素����、正式記錄的文件。CCS概念的提出說明對無菌產(chǎn)品生產(chǎn)污染的控制需要全生命周期的管理: 從項目的需求分析入手識別各維度污染風險; 設計污染控制策略��、配套控制措施來控制風險�����;在生產(chǎn)過程中進行污染的控制應對及做好日常監(jiān)測; 定期回顧評價污染控制策略的準確性和適應性��,及時進行動態(tài)調(diào)整修正��。
2.3 藥品質(zhì)量體系(PQS)
2022年版歐盟GMP附錄1 使用一個章節(jié)的篇幅(第3章藥品質(zhì)量體系) 強調(diào)適用于無菌產(chǎn)品的PQS的具體要求�����。中國GMP在總則中規(guī)定企業(yè)應當建立藥品質(zhì)量管理體系����,在附錄1中未針對無菌藥品提出藥品質(zhì)量體系的具體要求���。歐盟GMP 附錄1強調(diào)了無菌藥品生產(chǎn)的PQS應確保產(chǎn)品全生命周期的風險管理,對生產(chǎn)商���、高層管理人員���、負責無菌產(chǎn)品認證/放行的人員提出了具體要求,要對失敗進行根本原因分析���、CCS的開發(fā)和維護中施用風險管理����,涉及的最終處理�、貯存和運輸相關(guān)過程不應影響產(chǎn)品無菌、應在批次放行之前充分調(diào)查所有不合格等����。
2.4 屏障技術(shù)
2022年版歐盟GMP附錄1第四章“廠房”的“屏障技術(shù)”介紹了隔離器(isolator) 和限制進出隔離系統(tǒng)(restricted access barrier system,RABS) 兩種技術(shù)�����,并明確兩者是不同的技術(shù)。歐盟GMP規(guī)定隔離器“內(nèi)部工作區(qū)符合A級條件”�,中國GMP規(guī)定隔離操作器內(nèi)部環(huán)境為“B級(ISO 5級) 或更高潔凈度級別”,兩者對內(nèi)部環(huán)境級別要求存在差異�����。歐盟GMP附錄1明確RABS技術(shù)為“提供一個封閉但未完全密封�����、符合規(guī)定的空氣質(zhì)量條件的環(huán)境(對于無菌工藝為A級) 并使用剛性外殼和一體式手套使內(nèi)部與周圍潔凈室環(huán)境隔開的系統(tǒng)”��,中國GMP附錄1并未涉及RABS的相關(guān)規(guī)定����。此外歐盟GMP附錄1對隔離器和RABS手套的滅菌���、完整性測試頻次進行了明確的規(guī)定��,要求更為嚴格���。隔離器和RABS技術(shù)在無菌藥品的生產(chǎn)中應用廣泛,歐盟GMP對屏障技術(shù)的規(guī)定有利于促進屏障技術(shù)的規(guī)范應用����,降低技術(shù)應用中的無菌風險��。
2.5 潔凈室環(huán)境確認與監(jiān)測
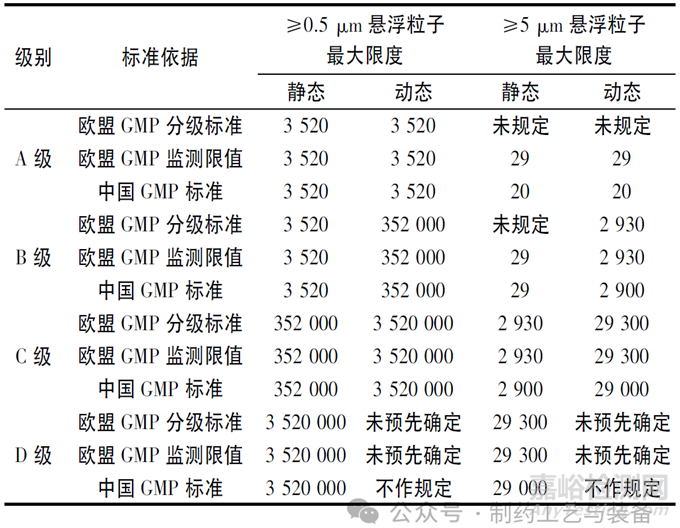
2022年版歐盟GMP附錄1將潔凈室確認(包括分級) 與動態(tài)環(huán)境監(jiān)測明確區(qū)別開來���,潔凈室確認標準在第四章“廠房”中進行了規(guī)定,環(huán)境監(jiān)測標準在第九章“環(huán)境監(jiān)測和工藝監(jiān)測”中進行了規(guī)定�����。潔凈室確認包含潔凈室分級�、微生物污染水平確認等,潔凈室分級的主要依據(jù)是空氣懸浮粒子標準�����。中國GMP附錄1未區(qū)分潔凈室確認(分級) 標準和環(huán)境監(jiān)測標準[7]��。
在空氣懸浮粒子方面����,歐盟GMP分級標準未規(guī)定≥5 μm懸浮粒子的A靜態(tài)和動態(tài)標準以及B級靜態(tài)標準,歐盟GMP環(huán)境監(jiān)測限值規(guī)定≥5 μm懸浮粒子的A靜態(tài)和動態(tài)現(xiàn)值為29個/立方米�����,中國GMP規(guī)定≥5 μm懸浮粒子的A靜態(tài)和動態(tài)標準為20個/立方米,這一點比歐盟GMP更為嚴格����。D級環(huán)境的動態(tài)懸浮粒子標準中國GMP為“不作規(guī)定”,歐盟GMP為“未預先確定”��,要求生產(chǎn)商根據(jù)風險評估和適用的常規(guī)數(shù)據(jù)建立動態(tài)限度����,標準制定更加科學。歐盟與中國GMP各級別空氣懸浮粒子標準對比見表2�。
表2 歐盟與中國GMP各級別空氣懸浮粒子標準對比(單位: 每立方米)
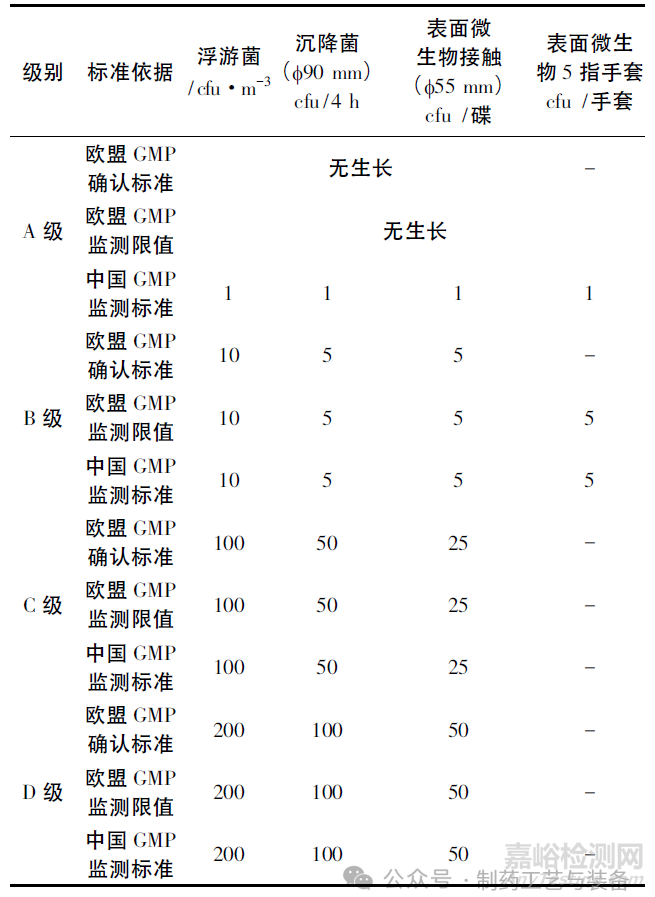
在微生物限度方面�����,在中國GMP 附錄1僅規(guī)定在動態(tài)環(huán)境監(jiān)測中進行微生物監(jiān)測����,歐盟GMP 附錄1要求在潔凈室確認以及環(huán)境監(jiān)測中均需要進行微生物監(jiān)測。中國GMP規(guī)定A級的微生物監(jiān)測的標準為<1 cfu��,此標準在執(zhí)行過程中存在如果環(huán)境監(jiān)測有微生物生產(chǎn)��,但監(jiān)測結(jié)果的平均值<1 cfu�����,檢測結(jié)果是否符合要求的爭議; 歐盟GMP將A級微生物標準修訂為“無生產(chǎn)”,消除了標準的異議����。歐盟與中國GMP各級別環(huán)境微生物限度標準對比見表3。
表3 歐盟與中國GMP各級別環(huán)境微生物標準對比
2.6 氣流流型測試
歐盟GMP 附錄1第四章對氣流流型提出了較高的要求���,條款“4.4”規(guī)定“應證明并確認整個A級區(qū)的單向流維護狀態(tài)”���,這就要求A級區(qū)域單向流的測試不能只測試某個位置,而應該證明整個A級區(qū)域內(nèi)部的單向流是符合要求的����。條款“4.15”規(guī)定“應該對潔凈區(qū)和潔凈室的氣流流型進行可視化研究,證明空氣沒有從較低級別區(qū)域進入到較高級別區(qū)域��,并且空氣不會從不太潔凈的區(qū)域(例如地板) 或經(jīng)過可能轉(zhuǎn)移污染的操作員或設備跨越到較高級別的區(qū)域”�,這就要求氣流流型測試不限于A級區(qū)域,不同級別潔凈區(qū)域間均需要測試�����,而且測試應包含靜態(tài)和動態(tài)條件。中國GMP附錄1僅規(guī)定“應當能夠證明所用氣流方式不會導致污染風險并有記錄(如煙霧試驗的錄像) ”��,對具體測試要求未進行詳細規(guī)定��。
2.7 液體過濾器的完整性測試
歐盟GMP附錄1條款8.87規(guī)定“應在使用前通過完整性測試(preuse post sterilization integrity test���,PUPSIT) 確認無菌過濾器組件的完整性��,以檢查由于使用前過濾器準備造成的損壞和完整性損失”“對液體進行除菌的除菌級過濾器�����,在使用后將濾器從殼體中取出之前應進行非破壞性的完整性測試”����。使用前滅菌后的完整性測試(PUPSIT) 要求非常高�,需要避免檢測時對已滅菌的過濾器系統(tǒng)造成二次污染?,F(xiàn)行中國GMP 附錄1對于液體過濾器的完整性檢測要求為“除菌過濾器使用后,必須采用適當?shù)姆椒⒓磳ζ渫暾赃M行檢查并記錄”�,未對PUPSIT提出要求[8-9]。
3����、討論
修訂后的2022年版歐盟GMP 附錄1要求更加嚴格、內(nèi)容更加全面�����、條款描述更加細致。
①適用范圍從“無菌藥品”放大到“無菌產(chǎn)品”����,涵蓋了無菌原料、輔料�、內(nèi)包裝材料以及最終成品制劑,強調(diào)無菌產(chǎn)品管理的目標不僅僅是微生物�,還包括微粒和內(nèi)毒素/熱原污染;
②引入了CCS的新概念,強調(diào)了QMS����、PQS 等理念;
③對無菌生產(chǎn)的具體技術(shù),如屏障技術(shù)(包括隔離器和RABS) ����、FFS、密閉系統(tǒng)���、SUS等�����,進行了詳細規(guī)定���,同時引入了快速轉(zhuǎn)移系統(tǒng)�、機器人�、快速微生物測試等新技術(shù);
④將潔凈室確認分級與動態(tài)環(huán)境監(jiān)測區(qū)別開來,各級別懸浮粒子和微生物標準設定更加科學;
⑤部分內(nèi)容要求比我國GMP更加嚴格����,如氣流流型測試范圍、液體除菌過濾器強調(diào)使用前滅菌后完整性測試(PUPSIT) 等���。我國GMP 附錄1的基本涵蓋了無菌藥品生產(chǎn)的關(guān)鍵要素���,但尚未明確PQS、CCS等理念����,缺乏對RABS、FFS�����、SUS等無菌生產(chǎn)技術(shù)的規(guī)定���。從無菌藥品生產(chǎn)理念和無菌藥品生產(chǎn)技術(shù)進步的角度來看�����,我國GMP 附錄1的修訂有其必要性�。2022年9月��,PIC/S根據(jù)已經(jīng)發(fā)布的2022年版歐盟GMP附錄1發(fā)布了自己的GMP指南附錄1��,而中國已經(jīng)在2021年提出加入PIC/S的申請�����,可以預見歐盟GMP附錄1 對中國制藥行業(yè)的影響也會不斷增強[10-11]�。在國際無菌藥品法規(guī)相互協(xié)同一致的背景下,我國監(jiān)管部門和無菌藥品生產(chǎn)企業(yè)要加快對歐盟GMP附錄1的研究和轉(zhuǎn)化���,及時修訂我國GMP附錄1���,促進我國無菌藥品質(zhì)量提升,建立國際化競爭優(yōu)勢�。
參考文獻
[1] EU.Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use,Annex1: Manufacture of Sterile Medicinal Products[EB/OL].[2022- 08 - 25]. https: / /health. ec. europa. eu /system/files /2022-08 /20220825_gmp-an1_en_0.pdf
[2] EU.Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use���,Annex1: Manufacture of Sterile Medicinal Products (corrected version) [EB/OL].[2008 - 12 - 15]. https: / /health. ec.europa.eu /system/files /2016-11 /2008_11_25_gmp-an1_en_0.pdf.
[3] 國家食品藥品監(jiān)督管理局.關(guān)于發(fā)布《藥品生產(chǎn)質(zhì)量管理規(guī)范( 2010 年修訂) 》無菌藥品等5個附錄的公告[EB/OL].[2011 - 02 - 24]. https: / /www. nmpa. gov.cn /xxgk /ggtg /qtggtg /20110224164501312.html
[4] 鄭金旺.2020 版歐盟GMP 附錄1 草案的主要變化解讀及對國內(nèi)無菌產(chǎn)品生產(chǎn)的影響分析[J].化工與醫(yī)藥工程�,2020,41(2) : 65-70.
[5] 曹鴻雁�����,韓瑩��,胡敬峰.山東省無菌制劑生產(chǎn)質(zhì)量風險分析與探討[J].中國藥事�,2018,32(7) : 901-905.
[6] 國家食品藥品監(jiān)督管理局藥品認證管理中心. 藥品GMP 指南: 無菌藥品[M]. 北京: 中國醫(yī)藥科技出版社�,2011: 118-123.
[7] 任杏珠.醫(yī)藥潔凈廠房空調(diào)系統(tǒng)確認和環(huán)境監(jiān)測取樣點選取探討[J].煤炭與化工,2022�����,45(1) : 144-150.
[8] 國家藥品監(jiān)督管理局.國家藥品監(jiān)督管理局關(guān)于發(fā)布除菌過濾技術(shù)及應用指南等3個指南的通告[EB/OL].[2018 - 09 - 11]. https: / /www. nmpa. gov. cn /ylqx /ylqxggtg /ylqxqtgg /20180911170301439.html.
[9] 胡敬峰�,許丹,韓瑩.除菌過濾技術(shù)在藥品生產(chǎn)應用中存在的問題與對策[J].中國藥事�,2020,34(12) : 1-5.
[10] PIC/S.Revised Annex 1 Manufacture of Sterile Medicinal Products[EB/OL].[2022- 09- 09]. https: / /picscheme.org /docview/4737.
[11] 國家藥品監(jiān)督管理局.國家藥監(jiān)局研究推進加入PIC/S工作[EB/OL].[2022 - 06 - 29]. https: / /www. nmpa.gov.cn /yaopin /ypjgdt /20220629093952150.html.
Critical zone – A location within the aseptic processing area in which product and critical surfaces are exposed to the environment.
關(guān)鍵區(qū):位于無菌工藝區(qū)內(nèi)的���、產(chǎn)品和關(guān)鍵表面暴露于其中的位置���。
Critical intervention – An intervention (corrective or inherent) into the critical zone.
關(guān)鍵干預:對關(guān)鍵區(qū)的干預 (糾正性或固有性干預)。
D-value – The value of a parameter of sterilisation (duration or absorbed dose) required to reduce the number of viable organisms to 10 per cent of the original number.
D 值:將活性微生物數(shù)量減少至原始數(shù)量的 10%的滅菌參數(shù)值 (持續(xù)時間或吸收劑量)��。
Dead leg – Length of non-circulating pipe (where fluid may remain static) that is greater than 3 internal pipe diameters.
死角:長度大于管道內(nèi)徑 3 倍的非循環(huán)管 (流體在此處可能保持靜止)�。
Decommission – When a process, equipment or cleanroom are closed and they will not be used again.
停用:工藝、設備或潔凈室關(guān)閉并且將不再使用��。
Decontamination – The overall process of removal or reduction of any contaminants (chemical, waste,residue or microorganisms) from an area, object, or person. The method of decontamination used (e.g.cleaning, disinfection, sterilisation) should be chosen and validated to achieve a level of cleanliness appropriate to the intended use of the item decontaminated. See also Bio-decontamination.
凈化:消除或減少區(qū)域����、物體或人體的任何污染物 (化學物質(zhì),) 廢物,殘留物或微生物) 的綜合過程。所用凈化方法 (例如清潔,消毒,滅菌) 應進行選擇和驗證,以達到適用于被凈化物品預期用途的潔凈水平另請參見生物凈化���。
Depyrogenation – A process designed to remove or inactivate pyrogenic material (e.g. endotoxin) to a specified minimum quantity.
除熱原:旨在將致熱物質(zhì) (例如內(nèi)毒素) 去除或滅活至規(guī)定最小量的過程���。
Disinfection – The process by which the reduction of the number of microorganisms is achieved by the irreversible action of a product on their structure or metabolism, to a level deemed to be appropriate for a defined purpose.
消毒:通過產(chǎn)品結(jié)構(gòu)或代謝的不可逆作用,將微生物數(shù)量減少至被認為適合某特定用途的水平的過程。
Endotoxin – A pyrogenic product (i.e. lipopolysaccharide) present in the Gram negative bacterial cell wall. Endotoxin can lead to reactions in patients receiving injections ranging from fever to death.
內(nèi)毒素:革蘭氏陰性細菌細胞壁中存在的致熱產(chǎn)物(即脂多糖)��。內(nèi)毒素能導致接受注射的患者發(fā)熱至死亡的反應��。
Equilibration time – Period which elapses between the attainment of the sterilisation temperature at the reference measurement point and the attainment of the sterilisation temperature at all points within the load.
平衡時間:從參考測量點達到滅菌溫度到負載內(nèi)所有點達到滅菌溫度之間的時間�����。
Extractables - Chemical entities that migrate from the surface of the process equipment, exposed to an appropriate solvent at extreme conditions, into the product or material being processed.
溶出物:當在極端條件下暴露于適當溶劑中����,從工藝設備表面遷移至被加工的產(chǎn)品或物料中的化學實體�。
First Air – Refers to filtered air that has not been interrupted prior to contacting exposed product and product contact surfaces with the potential to add contamination to the air prior to reaching the critical zone.
初始氣流:指在接觸暴露的產(chǎn)品和產(chǎn)品接觸表面之前沒有被阻礙從而在到達關(guān)鍵區(qū)之前不太可能被污染的經(jīng)過過濾的氣流���。
Filter Integrity test - A test to confirm that a filter (product, gas or HVAC filter) retain their retentive properties and have not been damaged during handling, installation or processing.
過濾器完整性測試:一種用以確認過濾器(產(chǎn)品���,氣體或 HVAC 過濾器)保持其截留特性并且在處理、安裝或加工過程中沒有被損壞的測試���。
本文作者胡敬峰���、明奕、王金子���、宋凱��、樊紅延���、馮巧巧,山東省食品藥品審評查驗中心����、國家藥品監(jiān)督管理局高級研修學院����,來源于藥學研究���。