基于2021年12月�,CFDI發(fā)布的《藥品注冊核查工作程序(試行)》等5個文件要求���,藥品注冊核查目的主要可概括為三個:①數(shù)據可靠性的核實和/或實地確證�����、②核實申報資料的真實性和一致性�����、③核實商業(yè)化的生產條件�����。包括研制現(xiàn)場核查和生產現(xiàn)場核查���,以及必要時的延伸檢查活動�。
研制現(xiàn)場核查是通過對藥品研制合規(guī)性�、數(shù)據可靠性進行檢查,對藥品注冊申請的研制情況進行核實�,對原始記錄和數(shù)據進行審查,確認申報資料真實性���、一致性的過程�����。研制現(xiàn)場核查的起點一般情況下為:確證性臨床試驗�����、生物等效性研究等藥物臨床試驗相關批次���,豁免藥物臨床試驗的�����,以進行質量對比研究的相關批次為起點���;未進行質量對比研究的�,以工藝處方基本確定后的批次為起點。終點為:商業(yè)規(guī)模生產工藝驗證批次前���。包括藥學研制現(xiàn)場核查���、藥理毒理學研究現(xiàn)場核查和藥物臨床試驗現(xiàn)場核查等;重點為:臨床試驗批次�、技術轉移批次、穩(wěn)定性試驗批次等影響藥品質量評價的關鍵批次�。必要時�,可前溯至研究立項�、處方篩選、工藝優(yōu)化等研究內容�。
藥學研制現(xiàn)場核查主要是對藥學研制情況,包括藥學處方與工藝研究�、樣品試制、質量控制研究及穩(wěn)定性研究等研制工作的原始數(shù)據�����、記錄和現(xiàn)場進行的核查�。
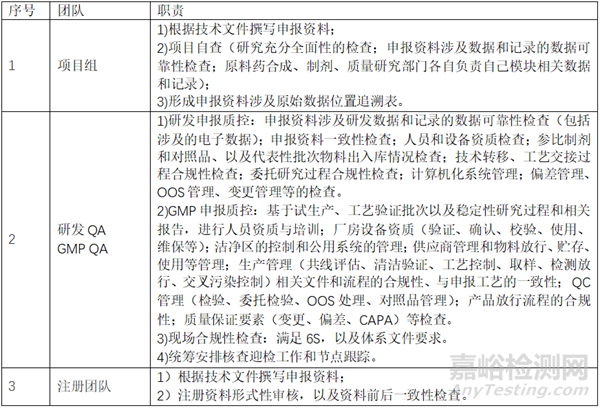
藥品注冊核查根據品種特性、被核查單位特點和風險���、審評過程意見���、有因檢查的原因等,確定核查內容�;各個公司職責劃分也不相同。本文就常規(guī)原料藥登記和制劑申報觸發(fā)的注冊現(xiàn)場核查�,藥學研制現(xiàn)場,研發(fā)團隊負責項目開發(fā)和分析方法驗證�����,GMP團隊負責試生產、工藝驗證���、穩(wěn)定性研究的情況���,相關迎檢準備工作和注意事項等介紹一二,以供大家參考�。
一、 工作職責劃分
建議在穩(wěn)定性3個月左右啟動項目自查工作�,項目組完成自查后,穩(wěn)定性4個月左右啟動QA的申報質控�。
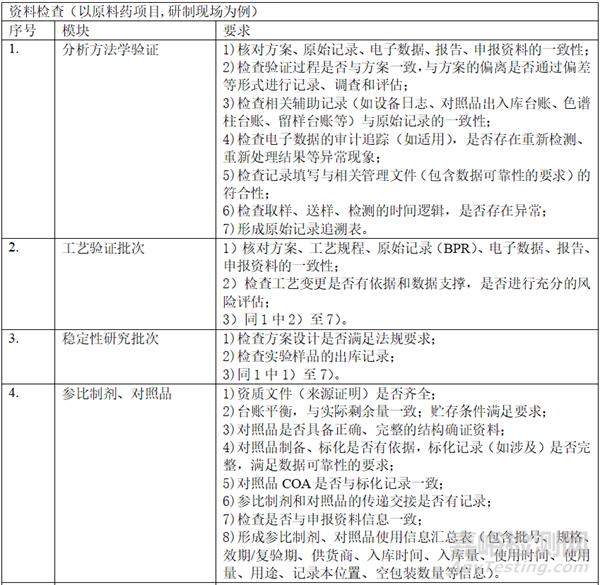
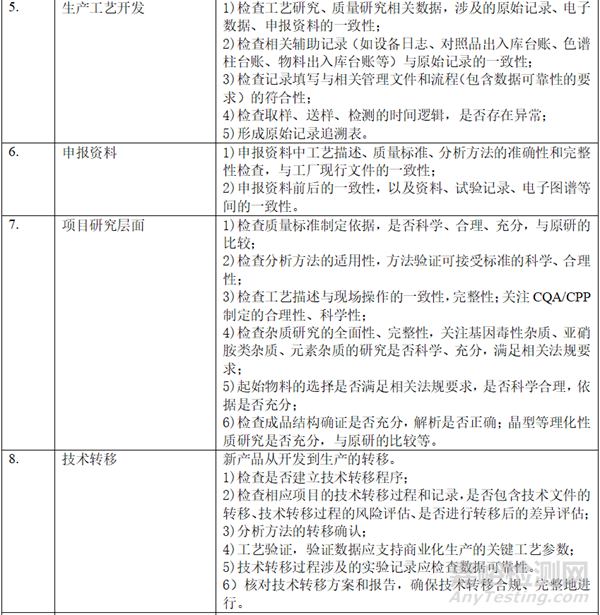
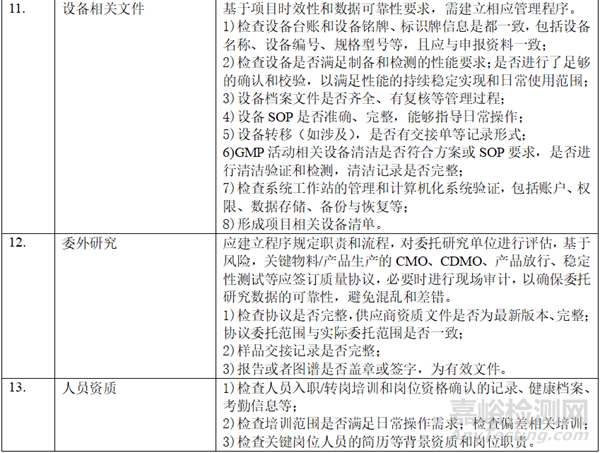
二、 資料準備
在自查的過程中���,形成以下表單:樣品試制清單(整體概況���,批次���、數(shù)量�����、時間�����、用途�����、剩余量)�����、研發(fā)和生產批次匯總信息表(代表性批次)�����、參比制劑使用信息梳理表(批次�、資質、出入庫數(shù)量和時間���、用途等)�����、對照品清單(批次���、資質�、出入庫數(shù)量和時間�、用途等)、項目相關研究設備清單�、項目相關檢驗設備清單、穩(wěn)定性研究清單�、研究人員清單、委托研究清單���。
三�����、 現(xiàn)場準備
按照6S要求進行準備和自查�����,需要注意控制混淆���、差錯、污染�����、交叉污染的風險���;關注物料標識�����、設備標識���、環(huán)境控制(如涉及)、人員防護裝備(實驗服���、手套���、口罩等)的穿戴、人員操作和記錄的及時性等�。
核查期間,每天上班之前�,可進行部門間的互查。
四���、 人員準備
需要就文件傳遞�����、問題回答���、現(xiàn)場介紹進行模擬演練�,確定相關問題的回答人員���,避免搶答�、緊張�����、職責不清等引起混亂�,并確保文件提供的準確性和及時性。