醫(yī)藥潔凈室空調(diào)系統(tǒng)應(yīng)能控制空氣污染�����,以保證藥品安全性����、有效性和質(zhì)量可控性。尤其作為確保臨床使用的無菌藥品生產(chǎn)的背景環(huán)境�,潔凈空調(diào)系統(tǒng)是無菌藥品生產(chǎn)企業(yè)的關(guān)鍵的公用系統(tǒng),藥品生產(chǎn)環(huán)境得到妥善的設(shè)計���、建造���、調(diào)試、運轉(zhuǎn)和維護����,則有助于確保產(chǎn)品的質(zhì)量[1]。中國藥品生產(chǎn)質(zhì)量管理規(guī)范(GMP) 規(guī)定�,應(yīng)當(dāng)根據(jù)藥品品種、生產(chǎn)操作要求及外部環(huán)境狀況等配置空調(diào)凈化系統(tǒng)�����,使生產(chǎn)區(qū)有效通風(fēng),并有溫度�、濕度控制和空氣凈化過濾,保證藥品的生產(chǎn)環(huán)境符合要求[1]�。2022年5月27日國家藥監(jiān)局發(fā)布中國GMP 臨床實驗用藥品附錄(2022年第43號) 公告,要求2022年7月1日起實施���,這是我國首次將臨床用藥納入GMP管理范疇����,附錄內(nèi)容規(guī)定:臨床試驗用藥品的制備和質(zhì)量控制應(yīng)當(dāng)遵循《藥品生產(chǎn)質(zhì)量管理規(guī)范》的相關(guān)基本原則及數(shù)據(jù)可靠性要求���,最大限度降低制備環(huán)節(jié)污染�����、交叉污染����、混淆和差錯的風(fēng)險�,確保臨床試驗用藥品質(zhì)量,保障受試者安全�����。制備臨床試驗用藥品的廠房、設(shè)施和設(shè)備應(yīng)當(dāng)符合《藥品生產(chǎn)質(zhì)量管理規(guī)范》及相關(guān)附錄的基本要求����。廠房、設(shè)施�����、設(shè)備的確認范圍應(yīng)當(dāng)基于風(fēng)險評估確定[2]���。
近年來,制藥行業(yè)的科技及技術(shù)在不斷進步�����,不僅在生產(chǎn)設(shè)備���、實驗室設(shè)備方面取得了進展���,而且在空調(diào)系統(tǒng)方面也取得了進展。歐盟GMP附錄1“無菌藥品生產(chǎn)”中明確提出�����,無菌藥品的生產(chǎn)應(yīng)符合特殊要求,以盡量降低微生物����、微粒和內(nèi)毒素/熱原污染的風(fēng)險。應(yīng)考慮以下關(guān)鍵領(lǐng)域應(yīng)按照GMP指南的相關(guān)章節(jié)對設(shè)施��、設(shè)備和工藝進行適當(dāng)?shù)脑O(shè)計����、確認和(或) 驗證,并在適用的情況下進行持續(xù)核實[3]���。持續(xù)有效的確認活動�,保障潔凈室潔凈級別持續(xù)符合產(chǎn)品及工藝需求�����,對實現(xiàn)向患者提供安全有效藥品的目標具有重要影響�。
1、潔凈室的確認法規(guī)要求
各國GMP對藥品生產(chǎn)用空調(diào)系統(tǒng)均有具體闡述�。中國GMP(第四章廠房與設(shè)施中規(guī)定) : 為降低污染和交叉污染的風(fēng)險,廠房����、生產(chǎn)設(shè)施和設(shè)備應(yīng)當(dāng)根據(jù)所生產(chǎn)藥品的特性���、工藝流程及相應(yīng)潔凈度級別要求合理設(shè)計、布局和使用�����,并符合下列要求�。應(yīng)當(dāng)根據(jù)藥品品種、生產(chǎn)操作要求及外部環(huán)境狀況等配置空調(diào)凈化系統(tǒng)�,使生產(chǎn)區(qū)有效通風(fēng)���,并有溫度�����、濕度控制和空氣凈化過濾�����,保證藥品的生產(chǎn)環(huán)境符合要求[2]�����。美國FDA“211.46通風(fēng)�、空氣過濾、空氣加熱與冷卻”[4]�����、歐盟GMP“3.12”章節(jié)����,以及WHO GMP通則和PIC /S GMP 指南的第一部分: 醫(yī)療產(chǎn)品的基本要求也有類似表述。
查閱各國GMP法規(guī)指南對潔凈室確認的相關(guān)要求����,中國GMP無菌附錄中僅簡單規(guī)定了風(fēng)速標準、單向流驗證的要求�����,也要求“根據(jù)潔凈度級別和空氣凈化系統(tǒng)確認的結(jié)果及風(fēng)險評估�,確定取樣點的位置并進行日常動態(tài)監(jiān)控”[2]。但未明確如何確認及驗證�����。歐盟GMP附錄一設(shè)置了專門的“潔凈室和潔凈空氣設(shè)備的確認”章節(jié)����,4.32~ 4.32共十項條款對確認形式�、項目等��,明確闡述了潔凈室和潔凈空氣設(shè)備的確認是評估分級的潔凈室或潔凈空氣設(shè)備與其預(yù)期用途的符合程度的整體過程�,應(yīng)根據(jù)所要求的環(huán)境特性進行確認,潔凈室確認(包括分級) 應(yīng)與動態(tài)環(huán)境監(jiān)測明確區(qū)別開來[3]���。GBT 25915.1-2021 潔凈室及相關(guān)受控環(huán)境�, ISO 14644-1: 2015 潔凈室及相關(guān)受控環(huán)境第一部分: 依據(jù)粒子濃度對空氣潔凈度的分級及GMP確認與驗證附錄提供了具體的實施指導(dǎo)����。
綜上可知,不論是美國���、歐盟還是中國,都從潔凈系統(tǒng)的設(shè)計和污染防控���、監(jiān)測等方面提出了系統(tǒng)的要求���。中國GMP和FDA均未規(guī)定如何進行潔凈室確認、確認哪些項目等���,僅歐盟GMP附錄一有詳細要求��,確認環(huán)境關(guān)鍵要素對維持無菌生產(chǎn)設(shè)施符合要求具有重要意義��。應(yīng)在整個設(shè)施內(nèi)實施污染控制策略(CCS) �,以界定所有關(guān)鍵控制點,并評估管理藥品質(zhì)量和安全風(fēng)險所用的所有控制措施(設(shè)計�����,程序性�����、技術(shù)性和組織性措施) 和監(jiān)測措施的有效性[3]���。
2��、潔凈室的確認流程
潔凈室的確認遵循中國GMP 確認與驗證的原則及基本要求���,應(yīng)建立確認與驗證的文件和記錄,并能以文件和記錄證明�����,并保持持續(xù)的驗證狀態(tài)。通常新建廠房設(shè)施較為復(fù)雜��,應(yīng)滿足GMP 要求����,也會運用項目管理等方法工具,會基于對產(chǎn)品和工藝的理解�,基于產(chǎn)品銷售市場的法規(guī)要求進行。其核心是界定系統(tǒng)及流程的所有關(guān)鍵控制點���,以及系統(tǒng)之間的相互作用�,對設(shè)施�、設(shè)備和生產(chǎn)工藝的風(fēng)險控制措施,并將所有關(guān)鍵控制點和控制措施的有效性進行關(guān)聯(lián)評估����。確保環(huán)境質(zhì)量持續(xù)滿足生產(chǎn)需求。為后續(xù)日常監(jiān)測以及相關(guān)控制方法提供依據(jù)�,確定日常監(jiān)控頻次。
潔凈室的確認和驗證方法: 通常在現(xiàn)場驗證主計劃中給出測定方法����,以及驗證策略���。應(yīng)用于確認的整個生命周期的系統(tǒng)的驗證方法�。潔凈室的確認通常涵蓋: 設(shè)計確認(DQ) 、安裝確認(IQ) ���、運行確認(OQ) ���、性能確認(PQ) 及再確認等幾個階段。應(yīng)定期評估持續(xù)保持驗證狀態(tài)��,當(dāng)發(fā)現(xiàn)趨勢出現(xiàn)漸進性變化時�,應(yīng)當(dāng)評估是否采取糾正措施,評估是否進行必要的再確認��。
由ISO14644 中可知����,在無菌藥品潔凈室確認的確認時,主要包括: 已安裝的過濾器系統(tǒng)的檢漏和完整性測試��、氣流測試(流速��、風(fēng)量) 及換氣次數(shù)����、自凈及恢復(fù)時間測試���、壓差測試、氣流方向測試和氣流流型研究���、溫度測定測試�、相對濕度測試���、懸浮粒子測定���、微生物污染監(jiān)測(浮游菌、沉降菌和表面微生物) 等��。潔凈室的確認目的是評估潔凈室的潔凈級別是否滿足其預(yù)期用途的過程��,通常會結(jié)合生產(chǎn)需求進行風(fēng)險評估����,測試時需考慮“靜態(tài)”測試及“動態(tài)”測試。在實際測試過程中�,設(shè)計確認應(yīng)當(dāng)證明設(shè)計符合用戶需求,并有相應(yīng)的文件�。安裝和運行確認完成并符合要求后��,方可進行性能確認[5]。
根據(jù)歐盟附錄一4.23用于無菌產(chǎn)品生產(chǎn)的潔凈室和潔凈空氣設(shè)備����,如單向流單元(UDAFs) 、RABS和隔離器�,應(yīng)根據(jù)所要求的環(huán)境特性進行確認。每個生產(chǎn)操作要求具有合適的動態(tài)下環(huán)境潔凈水平����,以最大程度降低所處理的產(chǎn)品或物料的污染風(fēng)險。應(yīng)維持“靜態(tài)”和“動態(tài)”下的適當(dāng)潔凈度水平���??芍F(xiàn)階段對無菌環(huán)境的要求更加的貼合實際情況來驗證��,要求相較更加嚴格�,覆蓋更加的廣泛,故而在實際操作中的動作影響應(yīng)多加進行風(fēng)險評估�����。
潔凈室的確認相對復(fù)雜���,影響因素較多���,結(jié)合法規(guī)要求及生產(chǎn)實際��,應(yīng)建立潔凈區(qū)性能確認管控策略�,下面舉例說明驗證確認中可能的問題及需關(guān)注要素����,見表1。
表1 潔凈室確認內(nèi)容及關(guān)注要素
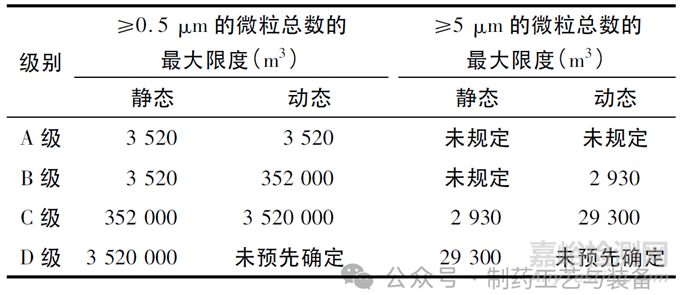
潔凈室確認(包括分級) 與動態(tài)的日常環(huán)境監(jiān)測應(yīng)在文件中明確區(qū)別并規(guī)定����。潔凈室分級是潔凈室確認的一部分,是一種根據(jù)潔凈室或潔凈空氣設(shè)備的標準通過測定總微粒濃度來評估空氣潔凈度水平的方法��。中國GMP無菌藥品附錄中�����,未明確潔凈室分級標準���。根據(jù)歐盟附錄一4.27對于潔凈室分級���,應(yīng)測定≥0.5 μm 和≥5 μm 的微?����?倲?shù)���。應(yīng)按照表2中潔凈室分級時各級別最大允許的總微粒濃度中規(guī)定的限度���,在靜態(tài)和模擬操作中都進行測定���。
表2 潔凈室分級與各級別最大允許的總微粒濃度
由表2可知,對A級≥5 μm 的微??倲?shù)與B級靜態(tài)的≥5 μm 的微粒總數(shù)要求更新未規(guī)定�,實際控制標準及要求無變化,僅根據(jù)在實踐過程中�,尤其是針對新建或改擴建廠房設(shè)施,應(yīng)當(dāng)對暴露的產(chǎn)品及部件足夠保護的狀況下���,監(jiān)測動態(tài)的實際意義不大����,基于風(fēng)險的簡化了驗證的流程�。在潔凈分級時����,應(yīng)在靜態(tài)和模擬操作中都進行測定���。
由表3可知����,對A級最大允許的微生物污染水平的要求更新為無生長��,該描述更加貼合實際����,更科學(xué)合理,也更能看出監(jiān)管機構(gòu)不認可平均后<1 的說法及做法�����。對未來的日常監(jiān)測及無菌理念有一定的正向影響�。
表3 確認過程中最大允許的微生物污染水平
3、潔凈室的確認實施問題思考
潔凈室的確認是一項綜合���、系統(tǒng)的���、復(fù)雜的工程��,會受到多種因素影響���,與廠房設(shè)施的設(shè)計、控制�、監(jiān)測等多維度、多因素有關(guān)���,如潔凈室的確認可能受各相關(guān)因素或系統(tǒng)影響[3]。在潔凈室確認實踐中�,經(jīng)常會發(fā)生不符合或偏差,應(yīng)在確認方案中明確偏差處理流程����,確保所有區(qū)域符合要求。常見問題及解決方法�,見表4。
表4 常見問題及解決方法
在歐盟附錄一發(fā)布后�����,氣流流型研究成為了業(yè)內(nèi)討論較多的話題���。(1) A 級單向流風(fēng)速測定: 在工作區(qū)域應(yīng)滿足0.36 ~ 0.54 m/s(指導(dǎo)值) 均勻風(fēng)速�,除非在CCS 中另有科學(xué)論證,氣流可視化研究應(yīng)與風(fēng)速測定相關(guān)聯(lián)[3]; (2) 氣流流型拍攝: 現(xiàn)階段對氣流流型研究的效果及目視觀察的形狀要求更高����,需在符合實際情況的動態(tài)操作下進行,且氣流流型清晰可見��,在拍攝途中可準備黑色幕布以保證清晰度�,同時需注意拍攝需有設(shè)備與拍攝目標的銘牌或編號,為確保數(shù)據(jù)可靠性有時會考慮采用三維視頻拍攝�。
4、小結(jié)
有效的無菌藥品潔凈室確認是質(zhì)量源于設(shè)計����、質(zhì)量源于控制策略的最佳實踐,潔凈室確認不是一次性確認����,應(yīng)基于法規(guī)要求及日常監(jiān)測趨勢預(yù)測變化,持續(xù)有效的確認是保障藥品無菌關(guān)鍵質(zhì)量屬性的前提及必要條件���。雖歐盟��、FDA和我國對無菌藥品及潔凈室確認的描述不盡相同�,但從其核心是以生產(chǎn)工藝需求的評估、設(shè)計��、確認����、監(jiān)測和控制為基礎(chǔ),基于對監(jiān)測數(shù)據(jù)所采集的信息及相關(guān)的特定GMP要求符合性的審核��,更全面�����、更準確進行產(chǎn)品無菌保證能力的評價����?��;陲L(fēng)險管理潔凈室全生命周期確認實施策略�����,為后續(xù)生產(chǎn)制造確定污染防控策略�,建立失敗的風(fēng)險發(fā)現(xiàn)機制�����。其目的是增加上市產(chǎn)品的無菌保障水平,確?��;颊哂盟幇踩?��,尤其針對避免處于臨床用藥期間的適用人群、受試者的風(fēng)險變得更為重要�。
參考文獻
[1] 冀紅. 論潔凈區(qū)( 室) 動態(tài)監(jiān)測中在線監(jiān)測系統(tǒng)的必要性[J]. 臨床醫(yī)藥文獻電子雜志, 2018�,5(30) : 184-185.
[2] 中華人民共和國衛(wèi)生部. 藥品生產(chǎn)和質(zhì)量管理規(guī)范( 2010 年修訂) [S]. 衛(wèi)生部令第79 號.
[3] European Commission. EU GMP Annex 1: Manufacture of Sterile Medicinal Products [EB /OL]. ( 2022-08-22) [2023-01-10]. https: / /health. ec. europa. eu /system/files /2022-08 /20220825 _ gmp-an1 _en_0. pdf.
[4] CFR 聯(lián)邦法規(guī): 第21 篇“食品和藥品”第211 部分制劑成品的現(xiàn)行生產(chǎn)質(zhì)量管理規(guī)范.
[5]ISO14644-1Cleanrooms and associated controlled environments Part1: Classification of air cleanliness by particle concentration.