2022年11月30日,NMPA發(fā)布“關(guān)于實施藥品注冊申請電子申報的公告(2022年第110號)���;公告指出��,自2023年1月1日起��,申請人提交的國家藥監(jiān)局審評審批藥品注冊申請以及審評過程中補充資料等����,調(diào)整為以電子形式提交申報資料���,申請人無需提交紙質(zhì)申報資料���,這意味著中國藥品注冊全面進入電子化;那么我們來梳理下電子通用技術(shù)文檔(eCTD)的發(fā)展史����,各國實施eCTD進展,以及我國藥品注冊電子化或eCTD實施進展;
電子通用技術(shù)文檔(eCTD)的發(fā)展史
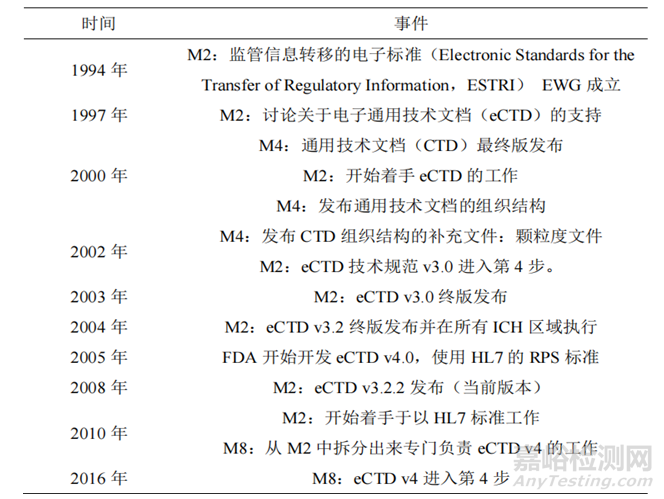
隨著經(jīng)濟全球化�,各國貿(mào)易往來頻繁,藥品也成為流通大環(huán)境的一員�;而各國的注冊監(jiān)管法規(guī)不同,企業(yè)要想在異國申報���,需要滿足不同監(jiān)管部門的要求��,這將會降低藥品上市速度并造成資源浪費�����;因此美國����、歐盟�、日本三方成員國組成了國際人用藥物注冊技術(shù)要求協(xié)會(ICH),在2000年�����,ICH開發(fā)出一套通用技術(shù)文檔(CTD)��,目的是為了提供一種全球統(tǒng)一的藥品注冊格式��,避免多國注冊時需要編寫不同注冊資料��。
2003年����,ICH M2提出第一版電子通用技術(shù)文檔eCTD V3.0(技術(shù)規(guī)范),它是通用技術(shù)文檔(CTD)的電子版�,該技術(shù)可進行全程化電子數(shù)字管理以及生命周期的管理,引入了MD5 checksum���,保證了遞交文件的安全性和保密性����。在2008年7月16日�,eCTD V3.2.2版本開始實施,2015年�,為了提高信息的穩(wěn)健性和靈活性,ICH發(fā)布了eCTD v4.0��,該版本能夠?qū)崿F(xiàn)信息雙通流向功能�����,V4.0支持所有申請之間的文件使用����,例如����,一個文件在 IND����、NDA中都需要遞交時,V4.0只需要遞交1次即可�����,引用其唯一ID來實現(xiàn)該文件重用��。2022年�,ICH開始大力推廣eCTD v4.0。截止目前���,大部分國家均處于從eCTD V3.2.2到eCTD v4.0的過渡階段���,其中歐洲、美國�、日本這三個國家預計在2026年、2028年���、2026年強制執(zhí)行eCTD v4.0��。
表1 eCTD發(fā)展史[1]
表2 eCTD v4.0的實施情況[2]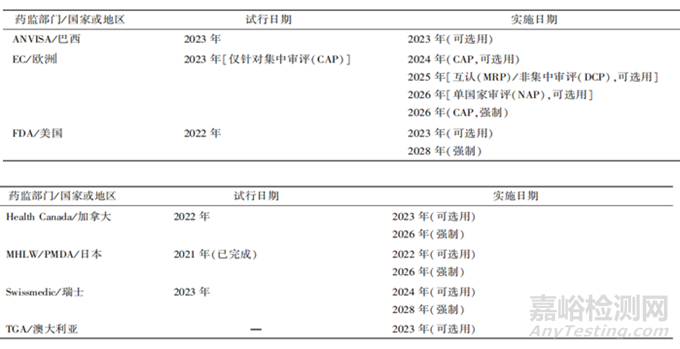
美國實施eCTD進展
在2001年時���,美國注冊申報方式主要是將CTD格式的注冊文檔打印下來,采用快遞形式進行運送����,但其存在的問題是效率低,審核困難��;為了提高審評效率�,縮短審評周期,2003年��,美國FDA開始接受使用eCTD V3.0 進行遞交��,2005年美國可接受的藥品注冊申請電子提交格式包括 e-NDA和e-ANDA格式( PDF文件)���、e-CTD 格式和NeeS格式等����。在2008年1月�,F(xiàn)DA規(guī)定e-CTD是唯一允許電子提交的格式�����,當然�,在當時FDA的紙質(zhì)檔案也是接受的���,也就是說���,2008年,F(xiàn)DA進入e-CTD和紙質(zhì)檔案共存過程����;這一過程持續(xù)了接近9年;在2017年5月15日起���,所有的新藥申請( NDA) ���、仿制藥申請( ANDA) 、生物制品申請(BLA) 及其補充申請必須以強制 e-CTD 格式提交�����;2018年5月5日��,F(xiàn)DA要求商業(yè)化IND和藥品主文件( DMF,3 類DMF除外)申報資料必須以 e-CTD 格式提交�,其他所有格式將被拒絕填寫,F(xiàn)DA在2018年全面進入e-CTD階段����。
圖1美國實行eCTD的發(fā)展史[3]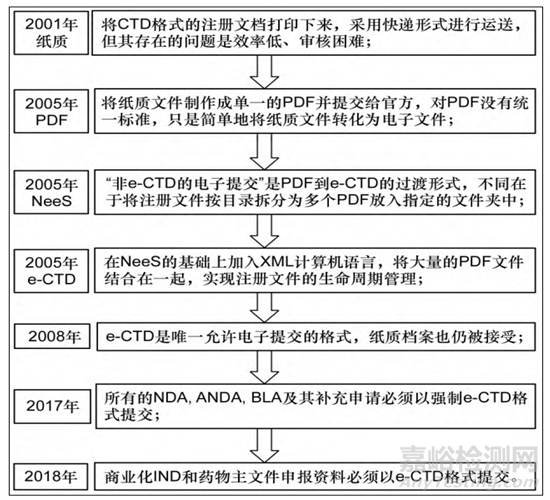
圖2 FDA申報流程圖[3]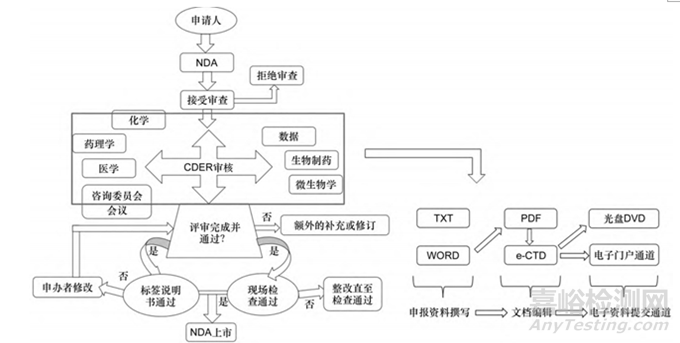
歐盟實施eCTD進展
歐盟在20世紀90年代就提出“電子提交”概念,如由德國開發(fā)的 “ DAMOS” 及法國開發(fā)的“MANSEV”均為歐盟國家的相關(guān)嘗試�����,ICH頒布的eCTD規(guī)范于2002年開始在歐盟施行�。2007年��,由于企業(yè)和審評機構(gòu)對于 eCTD 的使用率低下���,有一個變種的格式誕生了��,它遵循CTD 的結(jié)構(gòu)但是不支持生命周期管理�。它被命名為非 eCTD 電子提交(NeeS)格式�,是作為通往全面實行eCTD 之路的過渡形式,歐洲EMA則在2008年7月1日后接受單獨的eCTD格式注冊文件而不需再提交相關(guān)紙質(zhì)副本����,2018年起強制要求所有藥品上市都以eCTD格式申報��,不要求額外的紙質(zhì)文件����。2010 年7月1日開始�,對集中審評的人用藥物開始強制要求eCTD 格式。2015年7月1日開始�,非集中審評的新的申請開始強制實行eCTD 格式。2017 年1月1日開始�,互認可程序的新藥申請的提交強制實行eCTD 格式。
日本實施eCTD進展
日本藥品和醫(yī)療器械局(PMDA)于2004年開始要求使用eCTD�,2009年實施eCTDV3.2.2之后,接收到的eCTD 遞交數(shù)量急劇增加����,2015年12月后,日本的大部分新藥申請均以eCTD形式提交�����。
我國藥品申報電子化進展
相對于歐盟����、美國、日本我國藥品申報電子化進展相對較落后�����,起步較晚,在2004年���,我國首次提出綜述資料�����、質(zhì)量標準���、使用說明書和包裝標簽采用電子提交��,這也是我國電子申報的開端���,但僅限于部分資料�����,而且不強制����;
2008年05月07日�,為了提高審評效率�����,NMPA發(fā)布“關(guān)于通過中心網(wǎng)站提交藥品注冊申報資料電子文檔的通知”�,這是在《藥品注冊管理辦法(局令28號)》發(fā)布后����,NMPA啟動了過渡期品種的集中審評工作。通知要求:在CDE網(wǎng)站上傳docx格式的文件���,適用范圍:2007年10月1日之前已受理但尚未完成審評��,且之前未進行過電子提交的注冊申請��,2007年10月1日之后已受理��,但此前未進行過電子提交的注冊申請���。提交內(nèi)容包括質(zhì)量標準(草案)、藥品說明書����、立題目的與依據(jù)、對主要研究結(jié)果總結(jié)與評價、藥學研究綜述��、藥理毒理研究資料綜述��、綜述臨床試驗研究資料綜述等資料���;該通知的發(fā)布打開了我國藥品申報電子提交的開端�����。
2013年04月藥審中心將試點開展藥品注冊申報資料電子遞交工作�;2017年11月國家局發(fā)布“關(guān)于調(diào)整原料藥����、藥用輔料和藥包材審評審批事項的公告(2017年第146號)”公告提出:原料藥、藥用輔料和藥包材企業(yè)在藥審中心門戶網(wǎng)站“申請人之窗”填寫品種基本信息后�,將登記資料以光盤形式提交至藥審中心��;這是我國首次提出以光盤形式提交資料���,但目的僅用于現(xiàn)場核查檢驗�����;
2019年5月6日��,CDE發(fā)布“關(guān)于提交藥品注冊檢查檢驗用申報資料光盤的通知”�����,根據(jù)該通知的要求�,應(yīng)在受理后10日內(nèi)提交光盤,主要目的是方便現(xiàn)場檢查和檢驗����,而非用于受理或?qū)彶椋?/span>
2020年4月30日,CDE發(fā)布“公開征求《化學原料藥受理審查指南(征求意見稿)》意見的通知”���;2022年2月9日��,CDE發(fā)布“關(guān)于再次公開征求《化學原料藥受理審查指南(試行)(征求意見稿)》意見的通知”����,本通知中的受理審查指南是對“2017年第146號”公告的落實��,在“2017年第146號”公告中要求原輔包以光盤形式遞交申報資料����,雖然至今尚未轉(zhuǎn)正,但業(yè)界已經(jīng)按照最新征求意見的受理審查指南,開展原料藥的申報�����。
2022年1月29日���,CDE發(fā)布“關(guān)于疫情期間調(diào)整受理工作方式及接收申報資料要求的通知”����,為了控制日益增多的紙質(zhì)資料郵包帶來的疫情傳播風險���,CDE發(fā)布了該通知����。雖然本次通告并非eCTD申報�,但其提出了基于光盤資料進行受理審查的理念,也是我國藥品申報電子化進程中的重要節(jié)點���,不過這次要求受理后5個工作日提交紙質(zhì)資料��,也就是說這期間還是要求電子光盤+紙質(zhì)資料。
2022年11月30日�����,NMPA發(fā)布“關(guān)于實施藥品注冊申請電子申報的公告(2022年第110號公告;公告指出����,自2023年1月1日起,申請人提交的國家藥監(jiān)局審評審批藥品注冊申請以及審評過程中補充資料等��,調(diào)整為以電子形式提交申報資料����,申請人無需提交紙質(zhì)申報資料,申請人采用藥品電子通用技術(shù)文檔(eCTD)進行申報的��,無需再提交紙質(zhì)申報資料��,這意味著中國全面進入電子申報時代�,在這期間eCTD格式也是被認可。
圖3 2008年藥品審評中心電子提交系統(tǒng)[4]
圖4 我國電子申報發(fā)展史(注:紅色文字為eCTD相關(guān)的法規(guī)����,其他為電子注冊申報相關(guān))
綜上所述,中國的藥品注冊電子化主要經(jīng)歷如下��,①2004年部分資料電子化(綜述資料��、質(zhì)量標準等資料電子化);②2008年部分資料電子化(在2004年電子化的擴充)+紙質(zhì)資料提交����;③2017年,申報資料光盤提交主要用于現(xiàn)場核查��,而審查及申報資料與2008年(部分電子資料+紙質(zhì)資料)相同����;④2020年,原輔料申報采用電子申報(無紙質(zhì))�����,不過這次是征求意見稿�����,大部分企業(yè)按照征求意見稿實行原料藥申報����;⑤2022年,因為疫情申報資料電子光盤提交+紙質(zhì)資料�����;⑥2022年�,中國進入全面電子申報(光盤形式),無需提交紙質(zhì)申報��。
我國eCTD實施進展
2017年03月15日����,藥審中心開始加快推進eCTD項目建設(shè),2017年05月30日藥品審評中心發(fā)布關(guān)于征求《藥品電子通用技術(shù)文檔結(jié)構(gòu)(征求意見稿)》和《化學仿制藥電子通用技術(shù)文檔申報指導原則(征求意見稿)》意見的通知��,2017年10月再次發(fā)布關(guān)于藥品電子通用技術(shù)文檔結(jié)構(gòu)的征求意見稿�,直到2021年9月30日正式發(fā)出公告,國家藥監(jiān)局關(guān)于實施藥品電子通用技術(shù)文檔申報的公告(2021年第119號)����,實現(xiàn)藥品注冊申請的電子申報,提升“互聯(lián)網(wǎng)+藥品監(jiān)管”應(yīng)用服務(wù)水平�,國家藥品監(jiān)督管理局全面開展和推進了藥品電子通用技術(shù)文檔(eCTD)申報相關(guān)工作,實施范圍化學藥品注冊分類1類��、5.1類����,以及治療用生物制品1類和預防用生物制品1類的上市許可申請,可按照eCTD進行申報��,申請人應(yīng)按照eCTD技術(shù)文件(附件1-4)要求準備和提交eCTD申報資料光盤,并在eCTD注冊申請新報資料受理后5個工作日內(nèi)���,提交紙質(zhì)��;這是我國正式eCTD申報的開端����,不過��,這次eCTD并不夠徹底����,因為提交eCTD后還需要提交紙質(zhì)資料,而且僅針對部分��,提交的介質(zhì)為光盤����,而非網(wǎng)關(guān)至網(wǎng)關(guān)傳輸;并且非強制性要求���。
由此可見��,我國對于eCTD的目前處于選用階段�,僅僅是小范圍實施,主要還是以電子注冊為準��,eCTD不強求����,估計還有很多因素需要考慮�,特別是企業(yè)的響應(yīng)程度。
總結(jié):
從eCTD的發(fā)展史看��,我們得知���,eCTD的版本在不斷的升級����,管理和使用也是變得越來越方便��;從各國eCTD實施情況看�����,美國�、歐洲、日本實施eCTD較早��,大部分都經(jīng)歷過紙質(zhì)資料申報階段、紙質(zhì)資料+電子申報共存的過渡階段��、電子申報(格式多樣)�����、最后進入強制執(zhí)行eCTD階段���;而我國目前正進入電子申報+eCTD共存階段���,相信不久將來,我國可能也會與國際接軌��,完全進入eCTD階段�����。
以上是基于當前認知水平進行分析總結(jié)��,如有錯誤���,歡迎大家留言指正����。
參考文獻:
[1]呂婷等 美國和歐盟藥品申報電子提交的對比研究及對中國實施的啟示 2018 - 上海交通大學;
[2]陳 華等 我國正式實施 eCTD后藥品生產(chǎn)企業(yè)的注冊申報應(yīng)對策略 chinese journal of new drugs 2023 32(12)
[3]丁慧穎等 美國基于電子通用技術(shù)文件格式的藥品注冊申報制度研究及啟示 Chinese Journal of New Drugs 2023,32(21)
[4]NMPA官網(wǎng)
[5]李東昂等 我國藥品注冊申報資料實施eCTD格式的策略研究 中國制藥裝備·2018 年3 月·第 3 輯