[摘要] 本文基于對藥品注冊核查基本要求的思考與分析��,結(jié)合藥品注冊核查中關(guān)于真實(shí)性、一致性�、藥品上市商業(yè)化生產(chǎn)條件、研制的合規(guī)性和數(shù)據(jù)可靠性等核查內(nèi)容�����,以及藥理毒理學(xué)研究����、藥品臨床試驗(yàn)����、藥學(xué)研制及生產(chǎn)現(xiàn)場核查要點(diǎn)的研究����,從藥品注冊核查視角對藥品研制質(zhì)量管理中數(shù)據(jù)管理�、文件管理�、機(jī)構(gòu)與人員�、變更控制、偏差管理���、糾正與預(yù)防措施、委托管理���、生產(chǎn)管理�����、設(shè)施設(shè)備���、物料與樣品管理以及質(zhì)量控制和質(zhì)量風(fēng)險管理等要點(diǎn)進(jìn)行了識別與討論���,為進(jìn)一步做好藥品研制工作的質(zhì)量管理提供參考。
近年來�,根據(jù)黨中央國務(wù)院的要求�,結(jié)合行業(yè)及監(jiān)管發(fā)展的需要��,我國藥品相關(guān)法律法規(guī)進(jìn)一步得到優(yōu)化�����?����!吨腥A人民共和國藥品管理法》(2019 年修訂)及《中華人民共和國疫苗管理法》已于2019 年12 月1 日起實(shí)施,《藥品注冊管理辦法》���、《藥品生產(chǎn)監(jiān)督管理辦法》等一系列配套的法規(guī)文件也陸續(xù)頒布生效,持續(xù)深化藥品審評審批制度改革��,以臨床價值為導(dǎo)向���,鼓勵研究和創(chuàng)制新藥���,積極推動仿制藥發(fā)展,提升藥品質(zhì)量�,加快藥品上市速度��,更好地滿足人民群眾用藥需求���,建立了更全面�����、科學(xué)的藥品監(jiān)管體系與架構(gòu)。現(xiàn)有監(jiān)管框架中對藥品審評及核查工作有了新的要求與定位���,特別強(qiáng)調(diào)藥品研制和藥品注冊活動應(yīng)當(dāng)遵守有關(guān)法律���、法規(guī)、規(guī)章�、標(biāo)準(zhǔn)和規(guī)范����,應(yīng)當(dāng)保證全過程信息真實(shí)����、準(zhǔn)確、完整和可追溯�����。保證藥品研制工作符合當(dāng)前法律法規(guī)及技術(shù)指南的要求�,確保相關(guān)數(shù)據(jù)及資料的真實(shí)�����、充分��、可靠���,離不開覆蓋藥品研制過程的質(zhì)量管理體系���。近年來在藥品注冊核查中發(fā)現(xiàn)的問題也暴露出藥品研制階段質(zhì)量管理中存在的薄弱環(huán)節(jié)����。本文通過對藥品注冊核查的分析研究���,基于對藥品注冊核查要點(diǎn)的研究與思考,從藥品注冊核查的視角探討藥品研制質(zhì)量管理要點(diǎn)(主要包括以確證性臨床試驗(yàn)����、生物等效性研究等藥物臨床試驗(yàn)相關(guān)批次為起點(diǎn)�����,直至藥品注冊申請的商業(yè)規(guī)模生產(chǎn)工藝驗(yàn)證批次的藥品研制工作的質(zhì)量管理)��,以期為藥品研制單位提供參考,進(jìn)一步提高藥品研制與注冊的規(guī)范性與效率�。
1���、藥品注冊核查基本要求
藥品注冊核查是指為核實(shí)申報(bào)資料的真實(shí)性�、一致性以及藥品上市商業(yè)化生產(chǎn)條件��,檢查藥品研制的合規(guī)性����、數(shù)據(jù)可靠性等����,對研制現(xiàn)場和生產(chǎn)現(xiàn)場開展的核查活動以及必要時的延伸檢查活動[1] ,其核查內(nèi)容在一定程度上體現(xiàn)了當(dāng)前監(jiān)管機(jī)構(gòu)對藥品注冊研發(fā)質(zhì)量管理各要素的不同重視程度�����。目前���,國際上的批準(zhǔn)前檢查(pre?approval inspection�,PAI)通常是圍繞申報(bào)藥品對生產(chǎn)企業(yè)GMP 符合性進(jìn)行的檢查�,核心是圍繞產(chǎn)品檢查其GMP 符合情況和真實(shí)性[2] 。世界衛(wèi)生組織(Word Health Organization��,WHO) 認(rèn)為pai 的目的包括:評估企業(yè)是否符合GMP 要求(重點(diǎn)是環(huán)境����、質(zhì)量管理、人員���、設(shè)施和設(shè)備)����、評估產(chǎn)品生產(chǎn)與控制執(zhí)行的規(guī)程和申請材料的一致性��、審查注冊申請?zhí)峤毁Y料的完整性和準(zhǔn)確性��、批準(zhǔn)前生產(chǎn)批次與計(jì)劃進(jìn)行的商業(yè)批次的一致性及必要的抽樣[3] 。從定義上分析�����,我國的藥品注冊核查與目前國際上PAI的基本要求一致�����,但也存在一定差異���。
從藥品注冊核查的定位分析�����,藥品注冊核查是為保證藥品的安全、有效和質(zhì)量可控及鼓勵和推動藥品研發(fā)���、促進(jìn)制藥行業(yè)的健康規(guī)范發(fā)展�����、為藥品審評審批提供支持進(jìn)行的具有一定行政屬性的專業(yè)技術(shù)檢查���。從注冊核查方式分析��,藥品注冊核查通常采用現(xiàn)場核查的方式開展��,基于核查品種���、對象的特點(diǎn)及風(fēng)險等因素,可以采用多種模式與方法��;基于風(fēng)險研判���,部分研制工作可僅對記錄及數(shù)據(jù)等資料進(jìn)行核查��;根據(jù)實(shí)際情況及信息化管理手段的發(fā)展�,可以逐步探索采取遠(yuǎn)程核查等方式�����。從藥品注冊核查內(nèi)容分析,在《藥品注冊核查要點(diǎn)及判定原則》中分別針對藥理毒理學(xué)研究�、臨床試驗(yàn)�����、藥學(xué)研制和生產(chǎn)現(xiàn)場制定了對應(yīng)的核查要點(diǎn)����。核查內(nèi)容包括藥理毒理學(xué)研究原始資料�、生物等效性試驗(yàn)和藥物臨床試驗(yàn)研究原始資料、藥學(xué)研制原始資料�、申報(bào)品種的商業(yè)化生產(chǎn)條件和能力以及相關(guān)文件記錄。
整體上看,當(dāng)前法律法規(guī)框架下藥品注冊核查的任務(wù)發(fā)起、核查方式��、核查內(nèi)容及核查結(jié)果的處理均強(qiáng)調(diào)基于風(fēng)險���,其定位并不是上市前的GMP 符合性檢查���,核查方式主要采取現(xiàn)場核查的方式,其中部分內(nèi)容會采取基于記錄與數(shù)據(jù)審核的方式����,核查范圍包括藥理毒理學(xué)研究���、臨床試驗(yàn)�����、藥學(xué)研制和生產(chǎn)。與基于產(chǎn)品的上市前GMP 符合性檢查(pai)相比�,藥品注冊核查有其特殊性��,重點(diǎn)是為審評審批提供支持,從注冊核查的目的與要點(diǎn)角度分析,提示了當(dāng)前藥品研制階段質(zhì)量管理中一些應(yīng)重點(diǎn)注意的內(nèi)容�。
2、藥品注冊核查要點(diǎn)對藥品研制質(zhì)量管理的借鑒與思考
在理解藥品注冊核查的基本要求與特點(diǎn)的基礎(chǔ)上,通過對藥品注冊核查要點(diǎn)的分析,有助于從藥品注冊核查視角得出藥品研制質(zhì)量管理的要點(diǎn)���。藥品注冊核查內(nèi)容中��,真實(shí)性��、一致性、藥品上市商業(yè)化生產(chǎn)條件��、合規(guī)性與數(shù)據(jù)可靠性往往是相互關(guān)聯(lián)的�����,現(xiàn)場發(fā)現(xiàn)的同一個問題可能涉及核查要點(diǎn)的多個方面����。
2. 1 真實(shí)性
申報(bào)資料的真實(shí)性情況貫穿整個藥品注冊核查全過程��,在注冊核查涉及所有資料�、記錄、數(shù)據(jù)等內(nèi)容中均涉及真實(shí)性研判�����。《藥品管理法》��、《疫苗管理法》及《藥品注冊管理辦法》中針對提供虛假的證明����、數(shù)據(jù)、資料�����、樣品或者采取其他手段騙取臨床試驗(yàn)許可��、藥品注冊等許可的行為明確規(guī)定了處罰手段��。最高人民法院及最高人民檢察院聯(lián)合公布的《關(guān)于辦理藥品、醫(yī)療器械注冊申請材料造假刑事案件適用法律若干問題的解釋》中明確藥物非臨床研究機(jī)構(gòu)、藥物臨床試驗(yàn)機(jī)構(gòu)、合同研究組織的工作人員�����,故意提供虛假的藥物非臨床研究報(bào)告��、藥物臨床試驗(yàn)報(bào)告及相關(guān)材料�,應(yīng)認(rèn)定為刑法所規(guī)定的“故意提供虛假證明文件”?��!端幤飞a(chǎn)質(zhì)量管理規(guī)范》也明確禁止任何虛假、欺騙行為??梢哉f真實(shí)性問題是藥品上市許可持有人及有關(guān)單位進(jìn)行藥品研制��、生產(chǎn)、經(jīng)營與使用的最基本要求。
由于數(shù)據(jù)與記錄是藥品申報(bào)資料的基礎(chǔ)與來源�,也是藥品注冊申報(bào)相關(guān)單位開展各類藥品研制工作情況的基礎(chǔ)內(nèi)容與實(shí)際體現(xiàn)�����。在藥品注冊核查要點(diǎn)中,真實(shí)性方面明確提及的內(nèi)容集中體現(xiàn)為對數(shù)據(jù)與記錄真實(shí)性的核查�,藥品研制質(zhì)量管理最核心的內(nèi)容就是要保證關(guān)鍵研制數(shù)據(jù)與申報(bào)數(shù)據(jù)的真實(shí)性,保證相關(guān)數(shù)據(jù)與記錄的真實(shí),特別是杜絕惡意編造、偽造數(shù)據(jù)的情況,同時通過強(qiáng)化數(shù)據(jù)的規(guī)范性管理,降低真實(shí)性問題發(fā)生的風(fēng)險。
2. 2 一致性
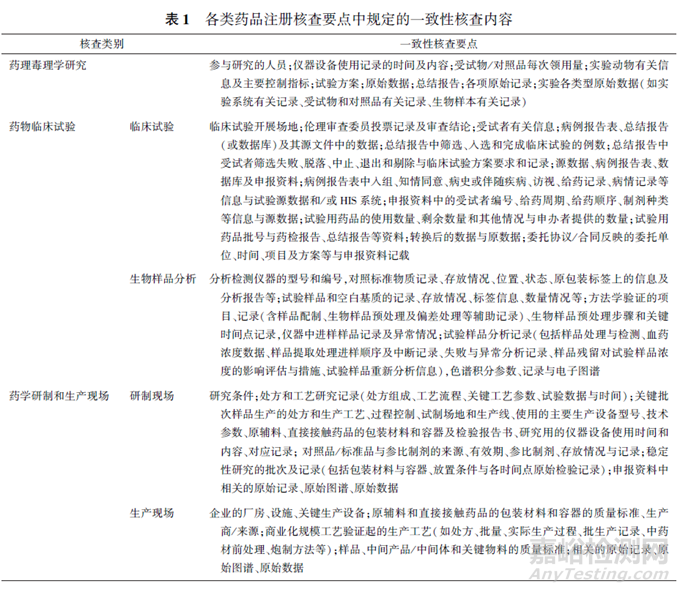
一致性的核查屬于對比性質(zhì)的核查�����,主要是申報(bào)資料與現(xiàn)場數(shù)據(jù)��、記錄�����、設(shè)施���、設(shè)備�����、人員�����、物料���、工藝等內(nèi)容的對比�。對藥品注冊核查中各類注冊現(xiàn)場核查要點(diǎn)(包括藥理毒理學(xué)研究[4] ���、藥物臨床試驗(yàn)[5] �����、藥學(xué)研制和生產(chǎn)現(xiàn)場[6])中明確描述需要核查一致性的內(nèi)容匯總分析見表1��。
在藥品注冊核查工作中�����,一致性的對比標(biāo)準(zhǔn)一般為申報(bào)資料(包括遞交的化學(xué)藥品生產(chǎn)工藝信息表、生物制品制造及檢定規(guī)程等)��。針對一致性的核查內(nèi)容不一定包括全部核查要點(diǎn)���,也不一定局限于核查要點(diǎn)所列內(nèi)容���,通常需基于風(fēng)險情況確定�����?��;谝恢滦砸蟮奶攸c(diǎn),其仍反映出數(shù)據(jù)管理是藥品研制質(zhì)量管理的核心內(nèi)容����,同時需要確保相關(guān)工作嚴(yán)格按方案規(guī)定執(zhí)行,并建立藥品注冊申報(bào)資料審核管理程序����,嚴(yán)格審核申報(bào)資料與實(shí)際數(shù)據(jù)的一致性。
2. 3 藥品上市商業(yè)化生產(chǎn)條件
針對藥品生產(chǎn)現(xiàn)場的注冊核查需要確認(rèn)生產(chǎn)企業(yè)是否具備藥品上市商業(yè)化生產(chǎn)條件����。從藥品質(zhì)量管理考慮,藥品質(zhì)量是設(shè)計(jì)出來的����,是生產(chǎn)出來的,也是管理出來的���。設(shè)計(jì)方面�,主要通過藥品注冊審評進(jìn)行研判;生產(chǎn)方面主要通過對影響相應(yīng)藥品質(zhì)量實(shí)現(xiàn)的要素進(jìn)行的審評及生產(chǎn)現(xiàn)場核查的研判���,即人�����、機(jī)����、料���、法�、環(huán)��、測等��;管理方面主要是對被核查單位藥品質(zhì)量管理體系中涉及對藥品關(guān)鍵批次研發(fā)及藥品生產(chǎn)上市放行前具有直接����、重大影響的質(zhì)量管理要素的現(xiàn)場核查�。
美國FDA 在CPGM 7346. 832 中闡述了PAI 關(guān)于商業(yè)化生產(chǎn)條件的5 個主要目的:① 生產(chǎn)與實(shí)驗(yàn)室的變化、調(diào)查和趨勢�����,產(chǎn)品生產(chǎn)證明該單位已對相關(guān)問題進(jìn)行了適當(dāng)評估。② 對組件�、過程控制材料、成品���、容器封蓋相關(guān)取樣�、測試和評估的合理程序與計(jì)劃�。③ 足夠的設(shè)施設(shè)備控制,有效防止交叉污染�����。④ 關(guān)于變更控制����、偏差調(diào)查、投訴與不良反應(yīng)處理��、召回����、相關(guān)異常情況報(bào)告監(jiān)管機(jī)構(gòu)等程序規(guī)定的充分性。⑤ 評估制定的商業(yè)化生產(chǎn)工藝和生產(chǎn)批記錄的可行性�,包括說明����、工藝參數(shù)和過程控制措施�����。
從保證藥品安全�����、有效及質(zhì)量可控角度分析��,結(jié)合藥品注冊核查的定位���,藥品上市商業(yè)化生產(chǎn)條件通常包括關(guān)鍵的藥品生產(chǎn)質(zhì)量實(shí)現(xiàn)要素及與藥品生產(chǎn)上市放行前具有直接���、重大影響的質(zhì)量保證要素。其中���,質(zhì)量實(shí)現(xiàn)要素包括圍繞申報(bào)產(chǎn)品的機(jī)構(gòu)與人員�、設(shè)備�、物料、生產(chǎn)工藝與操作、廠房設(shè)施���、質(zhì)量控制等方面(包括相應(yīng)的驗(yàn)證與確認(rèn));質(zhì)量保證要素包括與申報(bào)注冊藥品相關(guān)的文件化的質(zhì)量保證系統(tǒng)���、文件記錄管理�����、偏差管理���、變更控制、檢測超標(biāo)結(jié)果處理等內(nèi)容�。結(jié)合我國現(xiàn)行法規(guī)體系綜合考慮,其中部分內(nèi)容在藥品生產(chǎn)許可的檢查中進(jìn)行����;對于藥品生產(chǎn)質(zhì)量管理體系整體運(yùn)行的規(guī)范性,主要在上市前GMP 符合性檢查中進(jìn)行���。注冊核查對該部分的內(nèi)容主要圍繞產(chǎn)品生產(chǎn)工藝驗(yàn)證開展�����,重點(diǎn)是為藥品審評審批提供支持�����?����;谏鲜龇治?,藥品研制質(zhì)量管理中需要重點(diǎn)確保對應(yīng)設(shè)施設(shè)備、質(zhì)量管理體系�、數(shù)據(jù)與信息符合相應(yīng)的標(biāo)準(zhǔn)與要求,且需確保商業(yè)化生產(chǎn)條件的質(zhì)量管理基本與GMP 規(guī)定一致����。
2. 4 藥品研制的合規(guī)性
藥品研制的合規(guī)性主要指藥理毒理學(xué)研究工作是否符合《藥物非臨床研究質(zhì)量管理規(guī)范》(GLP)[7] 、生物等效性試驗(yàn)和藥物臨床試驗(yàn)研究是否符合《藥物臨床研究質(zhì)量管理規(guī)范》(GCP)[8] �����,藥品研制過程中的數(shù)據(jù)與記錄是否遵循《藥品記錄與數(shù)據(jù)管理要求(試行)》[9] ���,臨床試驗(yàn)用藥品的生產(chǎn)是否符合GMP 附錄《臨床試驗(yàn)用藥品(試行)》[10] �。藥品研制質(zhì)量管理應(yīng)確保各項(xiàng)工作的開展符合對應(yīng)的法規(guī)要求�����。
2. 5 藥品研制的數(shù)據(jù)可靠性
藥品研制的數(shù)據(jù)可靠性是指藥品研制數(shù)據(jù)完整、一致�����、準(zhǔn)確的程度[11-13] ���,包括藥品注冊相關(guān)電子數(shù)據(jù)與紙質(zhì)記錄。通過核查確認(rèn)藥品研制有關(guān)數(shù)據(jù)情況��,分析存在的數(shù)據(jù)可靠性問題���,評估相關(guān)數(shù)據(jù)對申報(bào)資料的影響程度���。數(shù)據(jù)是藥品質(zhì)量管理體系的基礎(chǔ)[14] ,是保證申報(bào)藥品安全����、有效和質(zhì)量可控的基礎(chǔ),藥品的注冊審批主要是基于申請人提交的數(shù)據(jù)開展的��。數(shù)據(jù)可靠性的核查貫穿于藥品注冊核查全過程�,所有對記錄、數(shù)據(jù)及關(guān)鍵計(jì)算機(jī)化系統(tǒng)的核查中都涉及對數(shù)據(jù)可靠性的基本分析,評估其真實(shí)���、準(zhǔn)確����、完整和可追溯的程度[9] ����。考慮到藥品研制數(shù)據(jù)管理存在數(shù)據(jù)量大��、來源廣泛����、類型多樣、跨越周期長�����、隨著研究的不斷深入對數(shù)據(jù)管理的要求程度不斷加強(qiáng)���、注冊申報(bào)數(shù)據(jù)僅為部分典型數(shù)據(jù)或代表性數(shù)據(jù)等特點(diǎn)���,其對數(shù)據(jù)管理與核查的要求與藥品生產(chǎn)和流通階段相比存在一定差異�,但核心內(nèi)容仍是“ALCOA + ”原則���,即可溯(attributable)�����、清晰(legible)��、同步(contemporaneous)、原始(original)�����、準(zhǔn)確(accurate)以及完整(complete)��、一致(consistent)����、持久(enduring)與可獲得(available)[15] ,藥品研制質(zhì)量管理應(yīng)基于風(fēng)險評估建立對不同階段數(shù)據(jù)管理的適宜要求與程序��。
3�、藥品研制質(zhì)量管理要點(diǎn)考慮
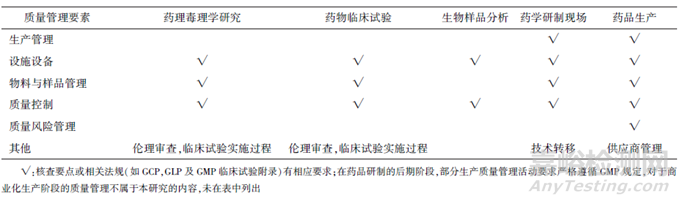
基于對藥品注冊核查基本要求及不同類型藥品研制現(xiàn)場核查要點(diǎn)的分析,結(jié)合藥品研制相關(guān)法規(guī)規(guī)定���,藥品研制工作質(zhì)量管理要點(diǎn)匯總情況見表2�。
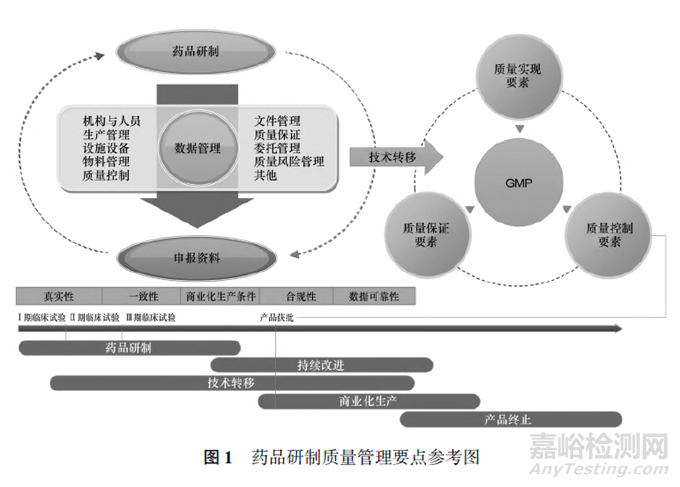
ICH Q10(藥品質(zhì)量體系)強(qiáng)調(diào)藥品質(zhì)量體系的設(shè)計(jì)需考慮到在藥品生命周期的各個階段不同目標(biāo)和可用的知識,應(yīng)考慮此項(xiàng)活動的規(guī)模和復(fù)雜程度��,其中:① 藥品研發(fā)活動的目標(biāo)是設(shè)計(jì)藥品及其生產(chǎn)工藝����,使其能始終如一地提供達(dá)到預(yù)期性能并滿足相關(guān)方需求的產(chǎn)品。② 藥品技術(shù)轉(zhuǎn)移的目標(biāo)是在研發(fā)和生產(chǎn)之間以及生產(chǎn)企業(yè)內(nèi)部或之間轉(zhuǎn)移產(chǎn)品和工藝知識來獲得要求的產(chǎn)品[16] �。故藥品研制質(zhì)量管理對不同研制階段可采取不同程度的要求,但對數(shù)據(jù)的管理將貫穿始終��,并作為核心要點(diǎn)����。由于藥品研制工作本身的特性以及對藥品研制從開發(fā)到商業(yè)化生產(chǎn)活動中遵循越來越高標(biāo)準(zhǔn)的要求,藥品研制質(zhì)量管理需要基于風(fēng)險評估來確定其各階段的要求��??偟膩碚f,需要建立一個質(zhì)量管理體系確保有足夠的資源(包括人員���、廠房���、設(shè)施����、設(shè)備�����、物料等)�����、組織機(jī)構(gòu)與文件化的程序要求�,遵循基于科學(xué)且基于風(fēng)險、持續(xù)改進(jìn)的原則開展藥品研制質(zhì)量管理(見圖1)�����。結(jié)合藥品注冊核查的關(guān)注點(diǎn)分析主要要點(diǎn)參考如下���。
3. 1 數(shù)據(jù)管理
數(shù)據(jù)(包括記錄)管理是藥品研制質(zhì)量管理最核心的內(nèi)容?��!秶宜幈O(jiān)局核查中心2021 年度藥品檢查工作報(bào)告》顯示在2021 年對1 214 個藥品注冊核查任務(wù)中發(fā)現(xiàn)的主要問題包括:① 藥理毒理學(xué)研究方面�����,如部分實(shí)驗(yàn)動物的檢測數(shù)據(jù)不一致�����,數(shù)據(jù)重測和結(jié)果取舍無相關(guān)標(biāo)準(zhǔn)操作規(guī)程����,數(shù)據(jù)處理執(zhí)行標(biāo)準(zhǔn)不一致,儀器使用和維護(hù)記錄��、受試物接收及運(yùn)輸記錄不完整等問題����。② 藥物臨床試驗(yàn)數(shù)據(jù)方面,如原始病歷記錄不詳細(xì)��、不完整���,方案偏離未報(bào)告�����,個別量表的填寫和修改不規(guī)范��,試驗(yàn)用藥的記錄不準(zhǔn)確�,安全性信息記錄不完整,合并用藥記錄不全等問題��。③ 藥學(xué)研制和生產(chǎn)現(xiàn)場方面��,如部分?jǐn)?shù)據(jù)存在無法溯源���、記錄不完整等數(shù)據(jù)可靠性問題,部分原始記錄與申報(bào)資料不一致����,技術(shù)轉(zhuǎn)移不充分�,確認(rèn)與驗(yàn)證不充分,不具備商業(yè)化生產(chǎn)條件等問題[17] ���。結(jié)合藥品注冊要求及注冊核查發(fā)現(xiàn)的典型性問題�,可以發(fā)現(xiàn)藥品研制環(huán)節(jié)質(zhì)量管理的核心在于數(shù)據(jù)管理,重點(diǎn)是保證研發(fā)數(shù)據(jù)的可靠性��,特別是對于可能對藥品安全性��、有效性和質(zhì)量可控性有影響的數(shù)據(jù)����,需要從系統(tǒng)角度建立有效的數(shù)據(jù)管理體系,確保數(shù)據(jù)符合“ALCOA+ ”原則���。
在注冊核查要點(diǎn)中就計(jì)算機(jī)化系統(tǒng)的驗(yàn)證與功能要求���、電子數(shù)據(jù)管理、紙質(zhì)記錄管理��、重點(diǎn)抽查的記錄與數(shù)據(jù)內(nèi)容等進(jìn)行了強(qiáng)調(diào)����。結(jié)合藥品研制工作質(zhì)量管理���,注意即使是早期的研究工作也應(yīng)做好記錄��,并對相應(yīng)數(shù)據(jù)進(jìn)行管理,避免丟失或刪除(特別是針對生產(chǎn)與檢測方面的內(nèi)容)�。同時不能忽視對申報(bào)資料的管理,申報(bào)資料應(yīng)根據(jù)其報(bào)告綜述類的特點(diǎn)�����,重點(diǎn)關(guān)注可溯、準(zhǔn)確��、完整�����、一致�����、持久與可獲得方面的要求����。需要理解做好數(shù)據(jù)的管理離不開系統(tǒng)的設(shè)計(jì)���、規(guī)劃與落實(shí),包括與數(shù)據(jù)可靠性相關(guān)的人員培訓(xùn)與要求����、制定并建立數(shù)據(jù)管理程序與措施(包括紙質(zhì)記錄和電子數(shù)據(jù))���、配備與所開展工作相適應(yīng)的計(jì)算機(jī)化系統(tǒng)與軟件并進(jìn)行適宜的驗(yàn)證�、建立并實(shí)施數(shù)據(jù)可靠性保證措施等方面��。
3. 2 文件管理
文件管理方面強(qiáng)調(diào)對所有質(zhì)量管理要求的制度化���,形成質(zhì)量管理文件,制定對應(yīng)的管理程序或標(biāo)準(zhǔn)操作規(guī)程(如物料管理程序��、生產(chǎn)與控制等程序���、質(zhì)量標(biāo)準(zhǔn)、設(shè)施設(shè)備組裝與清潔程序��、人員培訓(xùn)更衣與清潔程序���、分析儀器校準(zhǔn)程序���、質(zhì)量保證相關(guān)程序等)���。建立相應(yīng)文件管理程序保證各類管理程序及標(biāo)準(zhǔn)操作規(guī)程的有效性��、系統(tǒng)性����、協(xié)調(diào)性、可行性和可操作性[18] ����。
3. 3 機(jī)構(gòu)與人員
機(jī)構(gòu)與人員是保證藥品研制各個階段有效開展的基礎(chǔ)要素�,包括:倫理委員會、研究者���、臨床試驗(yàn)機(jī)構(gòu)���、申辦者、生產(chǎn)企業(yè)���、研制單位等,以及不同單位各自設(shè)置的部門與崗位(如監(jiān)查員����、稽查員、質(zhì)量負(fù)責(zé)人等)均需要建立明確的組織機(jī)構(gòu)��,規(guī)定各機(jī)構(gòu)、各部門及崗位的職責(zé)���,并確保配備具有適當(dāng)資質(zhì)(包括對質(zhì)量管理要求的理解與掌握)與適宜數(shù)量的人員���,制定文件化的培訓(xùn)程序規(guī)定與要求����。同時����,對于一些可能對產(chǎn)品質(zhì)量造成不利影響的行為建立相應(yīng)程序規(guī)定加以禁止���,如生產(chǎn)區(qū)域內(nèi)禁止吸煙����、飲食�����、種植植物等����。
3. 4 質(zhì)量保證
變更、偏差���、糾正與預(yù)防措施在藥品研制階段的要求隨著研制階段的逐步推進(jìn)有所區(qū)別,盡管基于為審評提供支持角度考慮�����,在藥理毒理學(xué)研究和臨床試驗(yàn)核查要點(diǎn)中沒有強(qiáng)調(diào)��,但其仍是藥品研制質(zhì)量管理的重要組成部分�����。變更是藥品研發(fā)過程的固有部分�����,其管理應(yīng)與藥品研發(fā)所處階段相適宜��,并應(yīng)進(jìn)行評估與記錄���。偏差對應(yīng)藥品研制的不同階段有著不同的理解與處理要求��,但任何階段對偏差的記錄與評估都有助于更好地理解產(chǎn)品與工藝�����。糾正與預(yù)防措施可以促進(jìn)質(zhì)量管理體系的持續(xù)改進(jìn)�����,并能實(shí)現(xiàn)藥品和工藝的改進(jìn)與更深入的理解����。
此外��,定期及專項(xiàng)自檢或來自外部的審計(jì)將有助于發(fā)現(xiàn)并控制風(fēng)險���,促進(jìn)質(zhì)量管理體系的持續(xù)改進(jìn)�����。
3. 5 委托管理
委托管理包括委托進(jìn)行藥理毒理學(xué)研究����、臨床試驗(yàn)、藥學(xué)研究�����、分析檢驗(yàn)及技術(shù)轉(zhuǎn)移的管理�����,應(yīng)當(dāng)對被委托單位的資質(zhì)予以確認(rèn)����,雙方簽署委托合同/ 協(xié)議���,明確雙方職責(zé)及有關(guān)要求��,允許委托方對被委托方進(jìn)行相關(guān)設(shè)施與活動的審計(jì)��,并加強(qiáng)對被委托單位數(shù)據(jù)可靠性的管控要求����。
3. 6 生產(chǎn)管理
生產(chǎn)管理方面��,研制現(xiàn)場主要關(guān)注關(guān)鍵批次樣品(如臨床試驗(yàn)批、生物等效性試驗(yàn)批和主要穩(wěn)定性試驗(yàn)批)的處方和生產(chǎn)工藝�����、過程控制��、試制場地和生產(chǎn)線��、使用的主要生產(chǎn)設(shè)備型號�����、技術(shù)參數(shù)及原始記錄等�。生產(chǎn)管理應(yīng)當(dāng)盡可能采取措施防止污染���、交叉污染以及混淆�����、差錯���,并隨著研制工作的推進(jìn),逐步識別��、確定關(guān)鍵質(zhì)量屬性和關(guān)鍵工藝參數(shù)�����,逐步建立明確的工藝參數(shù)及控制范圍�。注冊生產(chǎn)現(xiàn)場以商業(yè)規(guī)模生產(chǎn)工藝驗(yàn)證為起始,確認(rèn)企業(yè)生產(chǎn)工藝與注冊資料的一致性����,以及持續(xù)穩(wěn)定生產(chǎn)出符合注冊要求產(chǎn)品的能力,一般應(yīng)遵循商業(yè)化生產(chǎn)GMP 的有關(guān)要求��。
在藥品研制階段的生產(chǎn)管理中,需要注意知識管理及風(fēng)險管理的應(yīng)用�,一旦獲得足夠的數(shù)據(jù)需要對質(zhì)量屬性、關(guān)鍵質(zhì)量屬性���、工藝參數(shù)與關(guān)鍵工藝參數(shù)進(jìn)行定義與記錄����,注意保證相關(guān)數(shù)據(jù)符合“ALCOA+ ”原則�。
3. 7 設(shè)施設(shè)備
設(shè)施設(shè)備包括藥品研制所涉及各類設(shè)施(如實(shí)驗(yàn)室���、醫(yī)療急救設(shè)施�、生產(chǎn)廠房����、動物試驗(yàn)設(shè)施等)��、生產(chǎn)設(shè)備����、研究與分析儀器設(shè)備等,相關(guān)單位應(yīng)配備滿足研究需求的設(shè)施設(shè)備�,最大限度地降低混淆、差錯�����、污染與交叉污染的風(fēng)險�����,基于風(fēng)險原則做好校準(zhǔn)���、檢定、標(biāo)識、確認(rèn)����、使用���、清潔��、維護(hù)保養(yǎng)�����、維修等設(shè)施設(shè)備的生命周期管理工作,相關(guān)工作應(yīng)有適宜的記錄�,特別是與申報(bào)資料相關(guān)數(shù)據(jù)與記錄的可溯、一致�����、可獲得等要求��。此外�����,需做好臨床試驗(yàn)用藥共線生產(chǎn)的可行性與風(fēng)險評估和控制�,當(dāng)決定進(jìn)行共線生產(chǎn)時需確保對應(yīng)清潔程序的開發(fā)與驗(yàn)證的有效性。同時����,注意在生產(chǎn)區(qū)域內(nèi)不存儲和使用有毒有害物質(zhì)(如殺蟲劑����、有毒試劑等)。
3. 8 物料管理
樣品與物料管理包括對受試物/ 對照品��、試驗(yàn)用藥品��、對照藥品��、生物樣本/ 樣品����、對照標(biāo)準(zhǔn)物質(zhì)、試驗(yàn)樣品和空白基質(zhì)�����、對照品和參比制劑���、生產(chǎn)用物料(菌毒種���、細(xì)胞庫�、原輔料與直接接觸藥品的包裝材料和容器) 等采購自適宜的供應(yīng)商,并在各環(huán)節(jié)(如試制����、生產(chǎn)��、接收�、保存����、分發(fā)、使用����、留樣、返還或廢棄等)進(jìn)行的管控(包括標(biāo)識�、儲存條件與環(huán)境以及必要的隔離與進(jìn)入限制等),滿足其預(yù)期用途�����,確保相關(guān)記錄�、數(shù)據(jù)與證明材料的真實(shí)性及與申報(bào)資料的一致性,且符合數(shù)據(jù)可靠性要求�����。
3. 9 質(zhì)量控制
質(zhì)量控制包括藥品研制過程中涉及各類樣本分析檢測���,應(yīng)當(dāng)以分析檢驗(yàn)相關(guān)的人����、機(jī)、物����、法、環(huán)為核心�����,以檢驗(yàn)流程和檢驗(yàn)數(shù)據(jù)管理為基礎(chǔ)��,在遵循質(zhì)量管理有關(guān)內(nèi)容的前提下開展分析檢測工作(包括穩(wěn)定性試驗(yàn)檢測)[19] 。藥品注冊核查要點(diǎn)中根據(jù)研究項(xiàng)目的不同對此部分關(guān)注度較高的內(nèi)容包括:實(shí)驗(yàn)動物管理�����、對照品及標(biāo)準(zhǔn)物質(zhì)管理����、方法學(xué)驗(yàn)證�、分析檢測實(shí)施操作(如色譜積分等)、穩(wěn)定性研究�、原始記錄和檢驗(yàn)報(bào)告等�����。
藥品研制階段應(yīng)能確保提供足夠的資源開展質(zhì)量控制工作�����,其主要職責(zé)一般包括取樣與檢測����、執(zhí)行必要的確認(rèn)與驗(yàn)證���、標(biāo)準(zhǔn)物質(zhì)的管理��、執(zhí)行穩(wěn)定性試驗(yàn)計(jì)劃���、環(huán)境監(jiān)測以及分析方法驗(yàn)證�,特別注意對相關(guān)活動記錄與數(shù)據(jù)的保存��。
3. 10 質(zhì)量風(fēng)險管理
質(zhì)量風(fēng)險管理是藥品質(zhì)量管理必不可少的要素,可以促進(jìn)藥品質(zhì)量和工藝性能的持續(xù)改進(jìn)�����,盡管目前在藥品生產(chǎn)環(huán)節(jié)實(shí)際應(yīng)用較多�����,但其理念和方法應(yīng)貫穿藥品研制全過程��。藥品研制質(zhì)量管理中質(zhì)量風(fēng)險管理的目的是最小化研制工作內(nèi)在的風(fēng)險��,確保能基于科學(xué)與經(jīng)驗(yàn)識別并控制其中存在的風(fēng)險�����,其正式化程度和文件規(guī)范性要求可以隨著研制工作的推進(jìn)逐漸得到加強(qiáng)。
3. 11 技術(shù)轉(zhuǎn)移
在藥品研制階段的一些程序�����、方案�����、標(biāo)準(zhǔn)、工藝設(shè)計(jì)與驗(yàn)證���、分析方法等可能需要從研制場地轉(zhuǎn)移至商業(yè)化生產(chǎn)及檢測的場地���,對于此類技術(shù)轉(zhuǎn)移(包括產(chǎn)品���、工藝與知識的轉(zhuǎn)移)需要建立明確的程序進(jìn)行管理�,確保相關(guān)數(shù)據(jù)與記錄的詳細(xì)性、可追溯性和可獲得性處于適宜的程度����,保證轉(zhuǎn)移效果。具體在進(jìn)行技術(shù)轉(zhuǎn)移工作中可參考WHO及美國注射劑協(xié)會(PDA)針對技術(shù)轉(zhuǎn)移制定的技術(shù)指南[20-21] ����。
3. 12 其他
藥品注冊核查要點(diǎn)中針對不同類型研制工作描述的一些特定內(nèi)容也是藥品研制質(zhì)量管理的要點(diǎn)����,如倫理審查�、臨床試驗(yàn)實(shí)施過程、技術(shù)轉(zhuǎn)移和供應(yīng)商管理等���。同時�����,需要特別注意對生產(chǎn)過程中潛在污染的識別與控制(如消毒與衛(wèi)生)��,強(qiáng)調(diào)基于風(fēng)險確定相關(guān)驗(yàn)證與確認(rèn)的范圍與程度,包括工藝驗(yàn)證����、清潔驗(yàn)證���、分析方法驗(yàn)證等��,確保遵循了相應(yīng)的程序文件與方案�����,并保存好驗(yàn)證報(bào)告[22] 。
4��、結(jié)語
藥品質(zhì)量源于設(shè)計(jì)��,在藥品研制階段開展與對應(yīng)研制工作相適宜的質(zhì)量管理有助于進(jìn)一步滿足藥品研制工作對患者�、醫(yī)護(hù)人員��、監(jiān)管機(jī)構(gòu)及相關(guān)方的要求��。針對藥品研制工作的靈活性����、創(chuàng)新性等特點(diǎn),結(jié)合其數(shù)據(jù)及記錄在不同階段管理的特殊性��,以及各研究階段不同的特點(diǎn)與目標(biāo)要求�����,通過對質(zhì)量管理要點(diǎn)的識別與管控�����,構(gòu)建適宜的藥品研制質(zhì)量管理系統(tǒng)[23-24] ���,并持續(xù)改進(jìn)。減少藥品研制及注冊申報(bào)過程中在申報(bào)資料���、藥品上市商業(yè)化生產(chǎn)條件��、藥品研制的合規(guī)性、數(shù)據(jù)可靠性等方面出現(xiàn)的問題�����,有助于提高藥品及其工藝設(shè)計(jì)的充分性���,最大限度保障藥品研制的科學(xué)性與效率����。