關(guān)于首次人體試驗(yàn)的起始劑量設(shè)計(jì),F(xiàn)DA�、EMA�、ICH和NMPA都有相應(yīng)指南文件發(fā)布,各種文獻(xiàn)資料更是不勝枚舉����,但關(guān)于劑量爬坡階段的最高劑量設(shè)置依據(jù),可參考的文件卻鳳毛麟角�、鮮有提及。最近群里經(jīng)常有朋友提及這個(gè)話題���,大家也進(jìn)行了討論,一種觀點(diǎn)是參照動(dòng)物長(zhǎng)毒試驗(yàn)MTD的1/5-1/2,另外一種思路是動(dòng)物長(zhǎng)毒試驗(yàn)中引起中毒癥狀或臟器出現(xiàn)可逆性變化劑量的1/10�。當(dāng)然����,也可以參考同類藥物的文獻(xiàn)資料�。具體哪種說(shuō)法更為合理?有沒(méi)有監(jiān)管機(jī)構(gòu)明確的思路和建議����?
很高興此次跟趙老師一起合作���,對(duì)首次人體臨床研究的最高劑量設(shè)置依據(jù)進(jìn)行了梳理����,便于更進(jìn)一步、更充分的討論。但時(shí)間有限����,利用周末�,整篇文章從定題到完稿就用了1天多的時(shí)間,難免有紕漏,歡迎多提意見(jiàn)����。
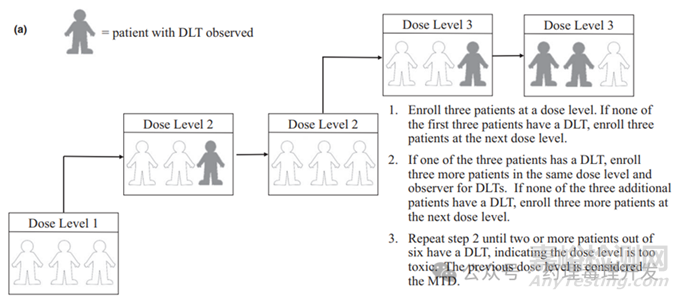
首先需要明確一點(diǎn),本次討論的話題是在臨床Ⅰ期試驗(yàn)啟動(dòng)前���,撰寫(xiě)臨床試驗(yàn)方案階段����,如何設(shè)置最高劑量,即pre-defined in the protocol����。實(shí)際的臨床最高劑量如最大耐受劑量(MTD)是按照類似下圖這種3+3設(shè)計(jì)或者通過(guò)PK/PD等分析手段研究出來(lái)的�。前者需要采用非臨床研究數(shù)據(jù)提前預(yù)測(cè),是IND之前需要完成的工作,后者則是根據(jù)臨床實(shí)測(cè)數(shù)據(jù)推導(dǎo),屬于臨床Ⅰa劑量爬升階段的工作�。
預(yù)設(shè)的首次人體試驗(yàn)最高劑量并不是一成不變的,而是根據(jù)臨床研究具體安全性表現(xiàn)或PD biomarkers,既可以提前終止�,也可以修訂方案繼續(xù)爬升至更高劑量。當(dāng)然�,第一步是要選擇合適的起始劑量計(jì)算方法獲得人體首個(gè)劑量,然后根據(jù)選定的劑量爬升策略進(jìn)行遞增�,以常見(jiàn)的3+3設(shè)計(jì)為例,按照改良的Fibonacci方法����,隨著劑量的增加����,遞增幅度越來(lái)越小�,通常依次按100%���、67%、50%����、40%和30%-35%遞增,一直遞增至預(yù)設(shè)的最高劑量����。所以這個(gè)預(yù)設(shè)的最高劑量通常怎么計(jì)算得到的呢?
先看下EMA相關(guān)資料����,2018年2月EMA發(fā)布了《Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products》����。指南中Section 7.5關(guān)于Maximum exposure and dose的原文如下:

EMA更多強(qiáng)調(diào)的是基于暴露量的概念����,臨床預(yù)設(shè)的最高暴露量需要結(jié)合所有已獲得的臨床前和臨床數(shù)據(jù),包括PD�、PK����、毒理綜合評(píng)估�。而且需要根據(jù)首次人體試驗(yàn)是健康人還是患者,分門(mén)別類的設(shè)計(jì)���。出于倫理考慮�,健康人所能接受的最大暴露量需要在藥效劑量范圍內(nèi),如果超出這一范圍,需要合理說(shuō)明���?��;颊邉t可以放寬要求���,探索MTD劑量�。其實(shí)在2018年�,EMA還發(fā)表了一篇文獻(xiàn)《Commentary on the EMA Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products》,對(duì)這一指導(dǎo)原則進(jìn)行補(bǔ)充說(shuō)明�,Dose selection, escalation steps and maximum exposure should consider the dose/exposure-response curves for relevant (desirable and adverse) effects, including the steepness and the plateau of the curve and the anticipated therapeutic dose. The reliability of these estimates may vary with the nature and novelty of the drug, the accuracy of biomarkers etc. 暴露量-有效性����、暴露量-安全性的關(guān)系�,量效關(guān)系是陡峭還是平坦���,有無(wú)可靠的biomarkers被重點(diǎn)提出�。
2019年�,Allergan、Roivant Sciences等藥企聯(lián)合發(fā)表的《Design and Conduct Considerations for First-in-Human Trials》一文中���,It is important to understand the therapeutic dose range and target saturation, as in most cases it is expected to predefine the expected maximum exposure level to be studied in an FIH trial in healthy volunteers. This is also discussed in the recent EMA guidance in which the maximum exposure in healthy volunteers should be within the estimated human pharmacodynamic range as an MTD approach is not appropriate. 文章中提及了治療劑量范圍����、靶點(diǎn)飽和及健康人臨床研究高劑量需在有效劑量范圍內(nèi)制定����,MTD不適用于健康人。并未提及針對(duì)患者的情況���。
結(jié)合EMA這篇指南以及工業(yè)界的解讀���,按照健康人和患者的情況,分別討論下����。首先對(duì)于健康人的計(jì)算思路還是比較清晰的���,非臨床基本都會(huì)開(kāi)展主要藥效學(xué)研究,或者PK/PD研究����,或者毒理研究中伴隨PD指標(biāo)考察(比如免疫檢查點(diǎn)抑制劑的受體占有率檢測(cè)),比較容易獲得暴露量的數(shù)據(jù)�。最大藥效劑量可以是腫瘤藥的腫瘤生長(zhǎng)抑制率,也可以對(duì)應(yīng)受體飽和這類PD指標(biāo)�,劑量進(jìn)一步提升已經(jīng)失去意義,那么對(duì)應(yīng)的暴露量可以用于健康人的最高劑量計(jì)算���。根據(jù)公式����,劑量可以通過(guò)AUC和CL獲得���,CL可以通常異速放大模型進(jìn)行種屬間換算(如通過(guò)食蟹猴的CL計(jì)算人體CL)����。當(dāng)然,這一暴露量在動(dòng)物毒理試驗(yàn)中應(yīng)該是NOAEL及以下劑量����,即有效且無(wú)毒����。
那么首次人體的目標(biāo)人群就是患者的情況,暴露量從何而來(lái)呢�,關(guān)于這點(diǎn),EMA并未提及文首提到的MTD的1/5-1/2的概念����,也未明確患者的具體算法,更談不上區(qū)分腫瘤患者和非腫瘤患者����。EMA更多是在保護(hù)健康人,對(duì)健康人的描述比較詳細(xì)���。對(duì)于患者雖沒(méi)有特別說(shuō)明�,但總體邏輯還是基于暴露量-安全性�、暴露量-有效性的關(guān)系,與健康人不同的是患者是要考慮臨床獲益的�,其中腫瘤患者與非腫瘤患者又有區(qū)別,前者屬于威脅生命的疾病,對(duì)安全性的包容度要高很多����。感覺(jué)上,EMA在疾病患者的臨床最大劑量設(shè)計(jì)方面�,把更多空間和自由度留給了企業(yè),是選用MTD的1/5-1/2���,還是非嚴(yán)重毒性反應(yīng)劑量����,或者最大PD效應(yīng)對(duì)應(yīng)的暴露量����,或許都是可以的。
解讀完EMA的思路����,再看下FDA的觀點(diǎn)。2019年FDA CDER抗感染產(chǎn)品部門(mén)的Ramya Gopinath博士在FDA Clinical Investigator Course培訓(xùn)中分享了《Safety Considerations in Phase Ⅰ Trials》����,其中關(guān)于最大劑量設(shè)定的建議如下圖所示,與EMA的觀點(diǎn)基本上是一致的�。不過(guò)�,從EMA和FDA的表述中�,能看到同一個(gè)關(guān)鍵詞“target saturation”,兩地監(jiān)管機(jī)構(gòu)均將這一PD指標(biāo)在最大劑量設(shè)計(jì)本身就資料寥寥����,提及的資料中也只言片語(yǔ)的情況下重點(diǎn)提出,更多是代表了一種態(tài)度����,PD指標(biāo)的最大效應(yīng)可作為最大劑量設(shè)計(jì)的關(guān)鍵考量依據(jù)�。如有可能,臨床前研究中盡量納入PK/PD的內(nèi)容����。

2014年4月,CDE張學(xué)輝�、卓宏、王濤等老師發(fā)表過(guò)一篇電子刊物《創(chuàng)新藥物臨床試驗(yàn)中暴露量-效應(yīng)關(guān)系研究的探討》����,其中關(guān)于首次人體試驗(yàn)劑量的選擇,表述如下:“首次人體試驗(yàn)是創(chuàng)新藥物研發(fā)過(guò)程中的重要里程碑之一�。在物種差異尚未完全明確的情況下,本試驗(yàn)是安全性風(fēng)險(xiǎn)最高的一個(gè)臨床試驗(yàn)�。因而在試驗(yàn)設(shè)計(jì)和具體實(shí)施上要格外慎重。最大推薦起始劑量的具體算法可參考國(guó)內(nèi)外相關(guān)的指導(dǎo)原則。最大劑量是根據(jù)動(dòng)物毒性試驗(yàn)的結(jié)果或同類產(chǎn)品應(yīng)用的劑量來(lái)確定���,應(yīng)起碼相當(dāng)于或略高于擬臨床常用劑量的高限�。”CDE這篇文章發(fā)布的時(shí)間比較早�,擬臨床常用劑量可以理解為最大或接近最大藥理活性劑量,比如臨床前受體飽和劑量����、最大激活劑量或信號(hào)通路完全抑制劑量等。這點(diǎn)與EMA����、FDA的思路有一些相像之處,只不過(guò)后來(lái)暴露量的概念逐漸得到重視����,使用頻率越來(lái)越高。
換個(gè)角度思考這個(gè)問(wèn)題���,為什么FDA�、EMA�、NMPA只有只言片語(yǔ)涉及到早期臨床方案中最高劑量設(shè)計(jì)?為什么對(duì)起始劑量的設(shè)計(jì)如此重視���?其實(shí)反過(guò)來(lái)思考這個(gè)問(wèn)題會(huì)發(fā)現(xiàn)一個(gè)淺顯易懂的結(jié)論����,起始劑量設(shè)計(jì)比最高劑量設(shè)計(jì)要重要很多。起始劑量確定以后����,根據(jù)產(chǎn)品的暴露量-安全性關(guān)系的陡峭程度、安全窗口或者毒性嚴(yán)重程度合理設(shè)計(jì)劑量爬升間距����。對(duì)于非臨床研究安全性表現(xiàn)好的產(chǎn)品如很多靶向藥���,可以適當(dāng)擴(kuò)大劑量間距比如2倍���,而且這類產(chǎn)品不一定非要探索MTD,其它終點(diǎn)比如最大可用劑量(MAD)也可以作為高劑量依據(jù)����。對(duì)于安全性風(fēng)險(xiǎn)高�、毒性大的產(chǎn)品���,可以設(shè)置比Fibonacci法更保守的劑間距�,高劑量可以采用劑量限制性毒性進(jìn)行約束���。所以,起始劑量是設(shè)計(jì)出來(lái)的�,最高劑量是根據(jù)臨床研究過(guò)程中的PK、PD���、安全性等數(shù)據(jù)試驗(yàn)出來(lái)的���。當(dāng)然,不是說(shuō)最高劑量的提前預(yù)設(shè)不重要或者沒(méi)有意義���,只是從監(jiān)管機(jī)構(gòu)和工業(yè)界的重視程度角度來(lái)看����,起始劑量更受重視�。
最后
無(wú)論如何,臨床方案需要提前設(shè)計(jì)����,最高劑量也需要有據(jù)可依���。結(jié)合各國(guó)監(jiān)管機(jī)構(gòu)和工業(yè)界零零散散的信息,可以得出以下幾點(diǎn)建議:
1) 非臨床研究設(shè)計(jì)時(shí)����,需充分重視PK(TK)/PD研究,盡量獲得充分的暴露量(Cmax���、AUC)數(shù)據(jù)?��?茖W(xué)合理選擇藥效模型�,重視臨床轉(zhuǎn)化���,如果能獲得可靠的PD biomarkers�,對(duì)于后續(xù)無(wú)論起始劑量還是最高劑量設(shè)計(jì)都非常重要���。
2) 暴露量-藥效(或PD)和暴露量-毒性關(guān)系的分析被屢次提及����,對(duì)于臨床最高劑量的設(shè)計(jì)至關(guān)重要���。健康人傾向于有效但無(wú)毒暴露量對(duì)應(yīng)的劑量����,患者可以略高于最大藥效劑量,但需要根據(jù)適應(yīng)癥合理評(píng)估毒性可接受程度�,并進(jìn)一步基于暴露量-毒性關(guān)系獲得最高劑量。需要注意的是�,建議選擇最敏感種屬或試驗(yàn)系統(tǒng)獲得的數(shù)據(jù)���。還要注意免疫激動(dòng)劑間的種屬差異比如因動(dòng)物和人體CD28受體豐度差異及起始劑量計(jì)算方法不合理導(dǎo)致的TGN1412慘痛經(jīng)歷���,這類產(chǎn)品高質(zhì)量����、來(lái)自人體樣本的體外藥理活性的數(shù)據(jù)對(duì)于起始和最大劑量的計(jì)算要給予足夠重視����。
3) 同類產(chǎn)品數(shù)據(jù)也很重要�,大部分產(chǎn)品其實(shí)都能找到相同靶點(diǎn)也好����,相似作用機(jī)制也好同類產(chǎn)品的文獻(xiàn)數(shù)據(jù)����,這些資料可以用來(lái)設(shè)計(jì)目標(biāo)產(chǎn)品的臨床最高劑量���。需要注意的是,不要照搬照抄���,結(jié)合產(chǎn)品之間活性����、藥效等方面的區(qū)別����,合理調(diào)整。
4) 藥品種類太過(guò)繁多���、復(fù)雜�,起始劑量的計(jì)算方法都不下十幾種����,最高劑量的獲取路徑也注定不會(huì)單一。根據(jù)目標(biāo)人群���、產(chǎn)品的類型����、同類產(chǎn)品可參考程度����、非臨床特點(diǎn)等�,有理有據(jù)����、合理設(shè)計(jì)。最高劑量設(shè)計(jì)過(guò)高則可能中途停止爬坡���,設(shè)計(jì)過(guò)低則需要修訂臨床方案����。
5) 由于人體研究的復(fù)雜性,首次人體試驗(yàn)中預(yù)設(shè)的最高劑量�,往往不一定和實(shí)際試驗(yàn)情況相符合,但最高劑量的限定原則往往是普遍適用的�。因此需要特別注意在臨床方案中將本品的最高劑量設(shè)定的考慮依據(jù)和原則寫(xiě)清楚���,這些原則既包括了對(duì)已有數(shù)據(jù)的考慮�,也需要基于對(duì)臨床中可能出現(xiàn)的情況做出假設(shè)并設(shè)定一些基于假設(shè)的考慮方向和原則���。方案中充分的闡述這些內(nèi)容�,有助于面對(duì)錯(cuò)綜復(fù)雜的實(shí)際情況����,當(dāng)臨床實(shí)際數(shù)據(jù)出現(xiàn)預(yù)期之外的情況時(shí),能夠很有效的基于預(yù)定的原則科學(xué)合理又能夠讓多方信服的去討論方案的修訂����,從而保障受試者權(quán)益的同時(shí)確保臨床研究的高效實(shí)施。