原料藥雜質(zhì)譜分析是藥物開發(fā)中的重要環(huán)節(jié)�,有利于了解藥物的安全性和有效性,有助于保證和提高藥品的質(zhì)量�。雜質(zhì)譜分析是雜質(zhì)研究工作的基礎(chǔ)環(huán)節(jié),全面的雜質(zhì)譜分析�,可為藥品制備工藝的開發(fā)和優(yōu)化、質(zhì)量控制策略的制定提供指導(dǎo)�,可使雜質(zhì)檢查工作有的放矢,是建立合理可行檢查方法的基礎(chǔ)�。筆者結(jié)合法規(guī)指南要求和自身經(jīng)驗(yàn)總結(jié)出以下五大方面雜質(zhì)譜分析的研究要點(diǎn):
一、 雜質(zhì)的來源及其控制策略
了解雜質(zhì)的來源�,可以幫助研究者減少或規(guī)避其產(chǎn)生。雜質(zhì)可能來自原料�、輔料、殘留溶劑�、生產(chǎn)工藝、包裝材料等�。因此,對(duì)制藥過程中的所有環(huán)節(jié)中雜質(zhì)的潛在來源之處全面分析�,并實(shí)施有效地控制是減少或避免雜質(zhì)產(chǎn)生的關(guān)鍵。根據(jù)《中國藥典》通則9102�,雜質(zhì)分為有機(jī)雜質(zhì)、無機(jī)雜質(zhì)�、殘留溶劑、異構(gòu)體雜質(zhì)、多晶型雜質(zhì)和基因毒性雜質(zhì)�,不同類型的雜質(zhì)來源略有不同,控制策略更是“因雜質(zhì)而異”�。以下是不同類型雜質(zhì)來源的分析和控制策略簡介:
1.1 有機(jī)雜質(zhì)
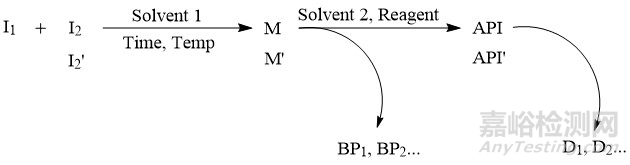
▲圖1-化學(xué)原料藥的合成工藝示意圖
原料藥中有機(jī)雜質(zhì)的來源往往通過合成工藝路線來綜合分析,比如圖1是一個(gè)化學(xué)原料藥的合成工藝示意圖�,根據(jù)此反應(yīng)路線,原料藥中有機(jī)雜質(zhì)的潛在來源主要包括以下幾個(gè)方面:①起始物料(I1�、I2)及中間體(M);②起始物料引入的雜質(zhì)(如I2′)�,以及起始物料引入雜質(zhì)的后續(xù)反應(yīng)產(chǎn)物,如I2′的后續(xù)反應(yīng)產(chǎn)物M′�、API′;③副產(chǎn)物�,如BP1、BP2等等�,如果副產(chǎn)物可隨主成分一同參與后續(xù)的反應(yīng),還需關(guān)注其后續(xù)反應(yīng)產(chǎn)物�;④原料藥的降解產(chǎn)物,如D1�、D2等等;⑤反應(yīng)中使用的試劑�、配位體、催化劑等等�。由于降解產(chǎn)物的化學(xué)結(jié)構(gòu)一般與活性成分類似或具有淵源關(guān)系�,通常也稱為有關(guān)物質(zhì)。除了降解產(chǎn)物外,其他4類有機(jī)雜質(zhì)都與制備工藝有關(guān)�,也稱為工藝雜質(zhì)。
1.1.1 工藝雜質(zhì)分析
(1)起始物料引入的雜質(zhì)及后續(xù)反應(yīng)產(chǎn)物
對(duì)于外購起始物料�,建議根據(jù)供應(yīng)商提供的制備工藝,對(duì)可能引入的雜質(zhì)進(jìn)行全面的分析和檢測(cè)�,注意分析起始物料引入雜質(zhì)在后續(xù)工藝步驟中的去向/清除情況,結(jié)合后續(xù)中間體中控?cái)?shù)據(jù)的積累�,合理制定起始物料引入雜質(zhì)的質(zhì)控策略(源頭控制,過程控制�,或在終產(chǎn)品中繼續(xù)關(guān)注)。
除起始物料引入雜質(zhì)外�,還建議重點(diǎn)關(guān)注那些可引入后續(xù)反應(yīng)的潛在雜質(zhì),通常這類雜質(zhì)的結(jié)構(gòu)和理化性質(zhì)與主成分類似�,可隨主成分一同進(jìn)行后續(xù)的化學(xué)反應(yīng),后續(xù)工藝步驟對(duì)其清除難度相對(duì)其他雜質(zhì)而言較高�,故在終產(chǎn)品中殘留的可能性也較大,這類雜質(zhì)可與有關(guān)物質(zhì)同時(shí)進(jìn)行檢查方法開發(fā)與驗(yàn)證�。
(2)副產(chǎn)物
建議根據(jù)工藝開發(fā)過程中掌握的工藝認(rèn)知、對(duì)所涉及化學(xué)反應(yīng)機(jī)理的理解以及數(shù)據(jù)的積累�,對(duì)各步驟可能產(chǎn)生的副產(chǎn)物進(jìn)行合理分析,并跟蹤其在后續(xù)工藝步驟中的去向/清除情況�,根據(jù)多批次跟蹤數(shù)據(jù)的積累,合理制定各工藝副產(chǎn)物雜質(zhì)的質(zhì)控策略�。同樣,建議重點(diǎn)關(guān)注與主成分結(jié)構(gòu)類似�、可引入后續(xù)反應(yīng)的副產(chǎn)物雜質(zhì)。
(3)反應(yīng)中使用的試劑、配位體�、催化劑等物料
當(dāng)某一種或多種雜質(zhì)排除為起始物料引入雜質(zhì)、副產(chǎn)物雜質(zhì)和降解產(chǎn)物雜質(zhì)后�,需要考慮是否為反應(yīng)中使用的試劑、配位體�、催化劑,或后處理中使用的樹脂�、吸附劑(如活性炭)等物料引入,建議對(duì)每一種物料進(jìn)行可能性分析�、逐一排查,當(dāng)確定雜質(zhì)由某種物料引入后�,應(yīng)當(dāng)評(píng)估是否更換甚至禁用該物料,必要時(shí)改變工藝�,以達(dá)到減少或清除該雜質(zhì)的目的。
1.1.2 有關(guān)物質(zhì)分析
建議可通過結(jié)構(gòu)特征的分析以及試驗(yàn)的手段來研究潛在的降解途徑和降解產(chǎn)物�,正式穩(wěn)定性試驗(yàn)、強(qiáng)制降解試驗(yàn)是常用的試驗(yàn)手段�。相對(duì)于正式穩(wěn)定性試驗(yàn),強(qiáng)制降解試驗(yàn)可在較短的時(shí)間內(nèi)獲得大量的有益信息�。因此在早期研發(fā)階段,強(qiáng)制降解試驗(yàn)是研究潛在降解途徑和降解產(chǎn)物的一種有效手段�。通過強(qiáng)制降解試驗(yàn),得出很多潛在的降解雜質(zhì)和降解途徑�,可為制定控制這些雜質(zhì)的策略提供有效的依據(jù),如工藝優(yōu)化�、選擇合適的儲(chǔ)存條件�。
1.2 無機(jī)雜質(zhì)
無機(jī)雜質(zhì)包括催化劑�、配位體和試劑�;重金屬元素雜質(zhì)(主要產(chǎn)生途徑為在藥物生產(chǎn)過程中接觸到的容器引入或者使用試劑和催化劑等時(shí)引入);無機(jī)鹽�;其他物質(zhì)(例如過濾助劑、活性炭等)�。其中重金屬元素雜質(zhì)為無機(jī)雜質(zhì)中最主要的雜質(zhì),本部分也將以此種類型雜質(zhì)為主進(jìn)行論述�。
1.2.1 元素雜質(zhì)的潛在來源
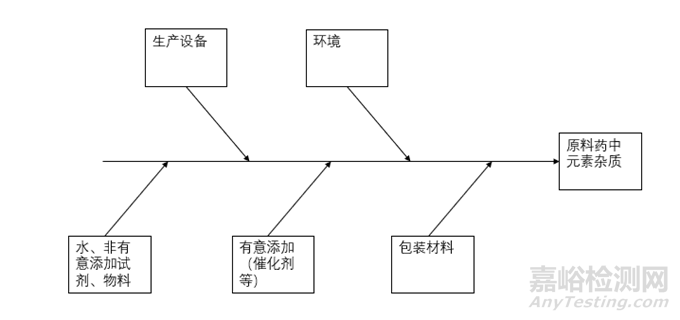
根據(jù)ICH Q3D,在藥品生產(chǎn)中�,元素雜質(zhì)的潛在來源廣泛,包括:
(1)在原料藥生產(chǎn)中有意添加元素(如催化劑)的殘留�;
(2)非有意添加但在原料藥生產(chǎn)所用起始物料、中間體�、水或其他試劑中可能存在的元素雜質(zhì);
(3)生產(chǎn)設(shè)備可能引入到原料藥中的元素雜質(zhì)�;
(4)包裝系統(tǒng)可能浸出至原料藥的元素雜質(zhì);
(5)環(huán)境中可能引入至原料藥中的元素雜質(zhì)�。
來源途徑可以由圖2簡單概括。通過對(duì)上述每一種潛在的來源進(jìn)行分析�,元素雜質(zhì)可以任何單獨(dú)或組合的形式被引入到藥品中。風(fēng)險(xiǎn)評(píng)估過程中�,元素雜質(zhì)的每一種來源的潛在貢獻(xiàn)都需考慮,以確定元素雜質(zhì)對(duì)藥品的整體貢獻(xiàn)情況�。
▲圖2-原料藥中元素雜質(zhì)潛在來源途徑示意圖
1.2.2 風(fēng)險(xiǎn)評(píng)估
ICH Q3D中表5.1列出了風(fēng)險(xiǎn)評(píng)估中建議考慮的元素雜質(zhì)�,共24種�。該表適用于藥品中所有來源的元素雜質(zhì)。該表中未列出的元素雜質(zhì)往往危害性較低�,在自然界中存在的含量較高,如鈉�、鉀、鈣�、鎂、磷�、硅、硼等�,通常不需進(jìn)行風(fēng)險(xiǎn)評(píng)估。
根據(jù)潛在元素雜質(zhì)的識(shí)別過程�,存在以下兩種可能結(jié)果:
(1)風(fēng)險(xiǎn)評(píng)估過程未識(shí)別出任何潛在的元素雜質(zhì)。應(yīng)記錄風(fēng)險(xiǎn)評(píng)估結(jié)論和支持性信息及數(shù)據(jù)�;
(2)風(fēng)險(xiǎn)評(píng)估過程識(shí)別出一個(gè)或多個(gè)潛在的元素雜質(zhì)。對(duì)于該過程中識(shí)別出的任何元素雜質(zhì)�,風(fēng)險(xiǎn)評(píng)估均需考慮元素雜質(zhì)來源多樣性,并記錄評(píng)估結(jié)論和支持性信息�。
原料藥、中間體�、起始物料、包裝系統(tǒng)和生產(chǎn)設(shè)備供應(yīng)商提供的關(guān)于潛在元素雜質(zhì)的信息有助于申請(qǐng)人的風(fēng)險(xiǎn)評(píng)估�。支持該風(fēng)險(xiǎn)評(píng)估的數(shù)據(jù)來源包括先驗(yàn)知識(shí)、公開發(fā)表的文獻(xiàn)�、相似工藝的數(shù)據(jù)�、供應(yīng)商信息或數(shù)據(jù)�、原料藥、中間體和起始物料的檢驗(yàn)數(shù)據(jù)等�。影響原料藥中潛在雜質(zhì)水平的因素也需在風(fēng)險(xiǎn)評(píng)估中予以考慮。這些因素包括在后續(xù)工藝過程中清除元素雜質(zhì)的有效性�、元素的天然豐度(對(duì)于非有意添加類的元素尤為重要)�、對(duì)于特定來源的元素雜質(zhì)濃度范圍的先驗(yàn)知識(shí)等。
1.2.3 控制
元素雜質(zhì)的控制是藥品整體控制策略的一部分�,用以確保元素雜質(zhì)不超過PDE 值(見ICH Q3D中表A.2.1,未列入表中的元素的PDE值可通過ICH Q3D中公式A.1.1計(jì)算或查閱相關(guān)法規(guī)�、文獻(xiàn))。當(dāng)元素雜質(zhì)水平超過控制閾值(PDE的30 %)時(shí)�,需采取額外的措施來確保元素雜質(zhì)水平不超過PDE值。常用的方法包括但不限于:
(1)調(diào)整生產(chǎn)工藝步驟�,通過特定或非特定的純化步驟將元素雜質(zhì)降低至控制閾值之下;
(2)實(shí)施工藝過程中或上游控制�,旨在將原料藥中元素雜質(zhì)的濃度限制在控制閾值以下;
(3)建立合成中間體的標(biāo)準(zhǔn)限度�;
(4)建立起始物料的標(biāo)準(zhǔn)限度。
各個(gè)元素雜質(zhì)的含量應(yīng)根據(jù) ICH Q6A 的原則進(jìn)行定期檢測(cè)�,通常每半年一次。
1.3 殘留溶劑
1.3.1引入途徑
原料藥中殘留溶劑的可能引入途徑主要有以下幾種:
(1)合成原料或反應(yīng)溶劑�;
(2)作為反應(yīng)副產(chǎn)物引入,如甲酯水解生產(chǎn)甲醇�;
(3)其他合成原料或其他溶劑帶入�,如甲苯�、苯胺中的少量苯;
(4)其他物質(zhì)(如大孔吸附樹脂中殘留的苯�、甲苯等)。
1.3.2 分類和限度
根據(jù)ICH Q3C�,殘留溶劑基于風(fēng)險(xiǎn)評(píng)估,按照對(duì)人體健康的潛在危害�,分為三類:
1 類溶劑:應(yīng)避免的溶劑
此類溶劑為已知的人體致癌物,強(qiáng)疑似人體致癌物�,以及環(huán)境危害物,共包括苯�、四氯化碳、1,2-二氯乙烷�、1,1-二氯乙烯、1,1,1-三氯乙烷五種�,1類溶劑均有明確的限度,必須嚴(yán)格控制在限度以下�。
2 類溶劑:應(yīng)限制的溶劑
此類溶劑為非遺傳毒性動(dòng)物致癌物,或可能導(dǎo)致其他不可逆毒性如神經(jīng)毒性或致畸性的溶劑�。可能有其他嚴(yán)重但可逆的毒性的溶劑�,共包括乙腈、氯苯�、氯仿等共31中溶劑。2類溶劑的限度可通過PDE法進(jìn)行計(jì)算�,計(jì)算方法參照ICH Q3C中3.3章節(jié)內(nèi)容�。
3 類溶劑:低潛在毒性的溶劑
此類溶劑為對(duì)人體低潛在毒性的溶劑�,包括乙酸、丙酮�、甲酸、乙醇�、乙酸乙酯等應(yīng)受GMP或其他質(zhì)量要求限制的常見溶劑(見ICH Q3C中表3)和石油醚、三氯乙酸�、三氟乙酸等無足夠毒理學(xué)數(shù)據(jù)的溶劑(見ICH Q3C中表4)。前者由于早已被公認(rèn)為低毒性�,對(duì)人類健康危害較低�,PDE值可認(rèn)定為≥50 mg/d,然后按照2類溶劑的計(jì)算方法得出其限度�;后者無PDE值,可通過公式ICH Q3C的附錄3中公式(1)計(jì)算得出�,然后同樣按照2類溶劑的計(jì)算方法得出其限度。
1.3.3 控制
若某種殘留溶劑超出限度�,主要的控制措施如下:
(1)優(yōu)化工藝,減少該溶劑用量�,想方設(shè)法控制在限度以下;
(2)改變工藝�,采用其他的替代溶劑或采用混合溶劑,如1類溶劑超出限度�,用2類或3類溶劑替代;2類溶劑超出限度�,用限度更寬的其他2類溶劑或3類溶劑替代�;3類溶劑超過限度�,用其他3類溶劑或水或有機(jī)/水混合溶劑替代;
(3)實(shí)施工藝過程中或上游控制�,旨在將原料藥中殘留溶劑的濃度限制在限度以下;
(4)制定合成中間體的殘留溶劑標(biāo)準(zhǔn)限度�;
(5)制定起始物料的殘留溶劑標(biāo)準(zhǔn)限度。
1.4 異構(gòu)體雜質(zhì)
異構(gòu)體雜質(zhì)一般在手性藥物中多見�,此類雜質(zhì)的研究是雜質(zhì)研究中的一個(gè)難點(diǎn)。根據(jù)《手性藥物質(zhì)量控制研究技術(shù)指導(dǎo)原則》報(bào)道�,異構(gòu)體雜質(zhì)的來源主要分為三種途徑:直接從起始原料或試劑中引入、不對(duì)稱合成和消旋體的拆分�。在手性藥物制備工藝研究中,如果能充分考慮從工藝中對(duì)產(chǎn)品的光學(xué)純度進(jìn)行有效的全程控制�,就能從源頭上控制產(chǎn)品的質(zhì)量。尤其是當(dāng)終產(chǎn)品難以全面有效地控制其光學(xué)純度時(shí)�,就更應(yīng)該重視制備工藝研究中的過程控制。
1.5 多晶型雜質(zhì)
原料藥的多晶型主要受不同析晶溶劑�、析晶溫度、析晶時(shí)間�、析晶操作條件影響。另外�,干燥、粉碎�、制粒、球磨等工藝步驟,溫度�、濕度、光照等環(huán)境因素的作用下�,可能出現(xiàn)轉(zhuǎn)晶現(xiàn)象,因此多晶型的雜質(zhì)來源較廣泛�。控制多晶型雜質(zhì)的措施主要還是從析晶工藝入手�。是否將多晶型檢查項(xiàng)訂入質(zhì)量標(biāo)準(zhǔn)可參考《化學(xué)仿制藥晶型研究技術(shù)指導(dǎo)原則(試行)》中決策樹1和決策樹2。
1.6 基因毒性雜質(zhì)
基因毒性雜質(zhì)的來源和控制策略與有機(jī)雜質(zhì)類似�,關(guān)鍵和前提是識(shí)別某雜質(zhì)是否為基因毒性雜質(zhì),然后采取對(duì)應(yīng)的控制措施�。具體識(shí)別和控制方法參考ICH M7中表1對(duì)此類雜質(zhì)的分類和控制。
二�、 雜質(zhì)的識(shí)別與定性
雜質(zhì)識(shí)別是指通過各種分析方法確定雜質(zhì)的化學(xué)結(jié)構(gòu),如結(jié)構(gòu)確證�。雜質(zhì)定性則是通過各類譜圖(如MS, NMR等)來推斷雜質(zhì)的結(jié)構(gòu)�、確定雜質(zhì)在譜圖中的位置和判斷原料藥中是否含有某種雜質(zhì)。各類雜質(zhì)的識(shí)別和定性方法如下:
(1)有機(jī)雜質(zhì)和基因毒性雜質(zhì)的識(shí)別通常用MS�、NMR、UV�、IR四大譜,有時(shí)將TGA�、DSC、有機(jī)元素分析法等作為輔助�;定性通常采用LC、GC、MS�、UV,其中基因毒性雜質(zhì)因限度很低(大多ppb級(jí))�,往往需要很強(qiáng)的靈敏度,故通常采用MS分析�;
(2)無機(jī)元素雜質(zhì)的識(shí)別和定性通常采用ICP-OES或ICP-MS,其中ICP-MS具有更高的靈敏度�、分辨力和專屬性;
(3)殘留溶劑通常采用GC和GC-MS�,對(duì)于苯、四氯化碳等限度較低的溶劑�,建議選擇靈敏度更高的GC-MS;
(4)手性雜質(zhì)的識(shí)別分為直接法和間接法:直接法指只需通過某單一方法即可確證手性藥物的構(gòu)型�,主要指單晶X射線衍射法(SXRD);間接法是指僅靠對(duì)待測(cè)物進(jìn)行分析�,尚難以確證其構(gòu)型,而需綜合其它數(shù)據(jù)�,如與其同系物的相關(guān)分析數(shù)據(jù)相結(jié)合才能確定待測(cè)物的構(gòu)型。如化學(xué)相關(guān)法�、比旋度、手性色譜�、核磁共振、旋光光譜(ORD)�、圓二色譜(CD)等。手性雜質(zhì)的定性方法主要包括比旋度�、使用手性色譜柱的高效液相色譜法(HPLC)或氣相色譜法(GC)�、化學(xué)相關(guān)法�;
(5)多晶型雜質(zhì)的識(shí)別和定性主要通過單晶X射線衍射法( SXRD)和粉末X射線衍射法(PXRD)。此外�,熱分析方法(如DSC、TGA和熱臺(tái)顯微鏡法等) �、光譜法(如IR、Raman和ssNMR等)�、 毛細(xì)管熔點(diǎn)法(MP)和偏光顯微法(PM)均可作為輔助手段進(jìn)一步支持不同晶型的確證。
三
雜質(zhì)譜的建立
基于對(duì)所有雜質(zhì)潛在來源的分析�、識(shí)別和定性后,針對(duì)每一種原料藥�,都應(yīng)建立一套完整的雜質(zhì)譜,包括所有可能的雜質(zhì)�,這需要一個(gè)大規(guī)模的研究工作。有了這個(gè)譜�,就可以在藥品生產(chǎn)過程中快速定性雜質(zhì),及時(shí)進(jìn)行控制�。
四、 定量分析及限度制定
雜質(zhì)的存在雖然難以避免�,但是必須限制其在藥品中的含量�。因此,需針對(duì)雜質(zhì)在產(chǎn)品中的最大限度�,開發(fā)出合適的定量分析方法。
4.1 限度制定
原料藥中有機(jī)雜質(zhì)的限度制定通常參考ICH Q3A中附件1的表格�,低于報(bào)告限的雜質(zhì)可不進(jìn)行控制;無機(jī)元素雜質(zhì)的限度制定通常參考ICH Q3D中的第7章內(nèi)容;殘留溶劑的限度制定通常參考ICH Q3C中第4章內(nèi)容�;基因毒性雜質(zhì)的限度通常參考ICH M7中第7章內(nèi)容或根據(jù)毒理學(xué)數(shù)據(jù)(如CPDB、Toxnet等)來制定�;異構(gòu)體雜質(zhì)和多晶型雜質(zhì)由于指導(dǎo)原則中并沒有明確的限度制定說明,需要研究者們查詢其他的文獻(xiàn)資料來制定�,如仿制藥的限度制定可參考已有的國家標(biāo)準(zhǔn)或國外標(biāo)準(zhǔn)或原研藥的質(zhì)量標(biāo)準(zhǔn);而對(duì)于創(chuàng)新藥�,就更需要研究者們多下功夫,若權(quán)威性的文獻(xiàn)資料查不到�,可在反復(fù)實(shí)驗(yàn)基礎(chǔ)上,依據(jù)經(jīng)驗(yàn)值來確定雜質(zhì)限度或者基于藥理毒理試驗(yàn)來確定雜質(zhì)限度�。
4.2 定量分析方法
雜質(zhì)的定量分析方法用于準(zhǔn)確、及時(shí)地監(jiān)測(cè)藥品中雜質(zhì)的含量�,以便必要時(shí)采取控制措施。通常定量分析方法與定性分析方法一致�,關(guān)鍵是根據(jù)雜質(zhì)限度和重要程度(如存在的可能性、清除難易�、是否為特定雜質(zhì))選取最準(zhǔn)確的定量方式。常用的定量方式包括峰面積歸一化法�、外標(biāo)法、主成分自身對(duì)照法�、內(nèi)標(biāo)法、標(biāo)準(zhǔn)加入法等等�。
五、 方法驗(yàn)證與穩(wěn)定性研究
開發(fā)出的定量分析方法需要進(jìn)行驗(yàn)證�,以確保方法的可靠性�。另外�,還需要研究原料藥的穩(wěn)定性,以便了解在藥品貯存期間�,雜質(zhì)的變化情況。
方法驗(yàn)證通常參考《中國藥典》指導(dǎo)原則9101�、USP<1225>、ICH Q2等指南�,雜質(zhì)的主要驗(yàn)證項(xiàng)目有專屬性、精密度�、準(zhǔn)確度、檢測(cè)限/定量限�、耐用性等。另外�,元素雜質(zhì)由于其特殊性,應(yīng)以USP<233>或EP 2.4.20為主進(jìn)行方法驗(yàn)證�。
方法驗(yàn)證結(jié)束后,該方法將一直用于工藝驗(yàn)證和穩(wěn)定性研究期間雜質(zhì)的檢測(cè)�。穩(wěn)定性研究主要參考ICH Q1和《中國藥典》指導(dǎo)原則9001進(jìn)行,根據(jù)指南要求�,應(yīng)當(dāng)首先進(jìn)行影響因素試驗(yàn),以便了解影響雜質(zhì)穩(wěn)定性的因素及可能的降解途徑與降解產(chǎn)物�,為生產(chǎn)工藝、貯存條件和是否需要變更降解產(chǎn)物分析方法提供科學(xué)依據(jù)�,也為后續(xù)的加速試驗(yàn)和長期試驗(yàn)條件提供參考�。而加速試驗(yàn)和長期試驗(yàn)的數(shù)據(jù)將直接為原料藥的貯存�、運(yùn)輸條件提供科學(xué)依據(jù)�。
小結(jié)
雜質(zhì)研究是貫穿于藥品研發(fā)始終的一項(xiàng)重要內(nèi)容,直接影響藥品的有效性和安全性�,雜質(zhì)譜分析是雜質(zhì)研究工作的基礎(chǔ),基于雜質(zhì)譜分析的雜質(zhì)控制是“質(zhì)量源于設(shè)計(jì)”基本理念在雜質(zhì)研究與控制中的一種具體實(shí)踐�。合理的雜質(zhì)譜分析策略有助于節(jié)省研究時(shí)間和全面地分析雜質(zhì),更有利于藥品的成功注冊(cè)申報(bào)�。通過本文的討論,筆者希望給廣大同行們一些有益的啟示—如何從雜質(zhì)譜分析入手確立科學(xué)的雜質(zhì)研究思路�。