近年來,在材料科學����、生物工程和信息技術等領域的進步推動下��,我國醫(yī)療器械的創(chuàng)新能力不斷增強,產(chǎn)業(yè)發(fā)展規(guī)模迅速擴大�����。為更好地適應產(chǎn)業(yè)發(fā)展趨勢和需求�����,我國監(jiān)管部門持續(xù)深化審評制度改革���。醫(yī)療器械的分類管理作為一項重要基礎性制度��,關乎著審評資源與監(jiān)管資源的分配����。如何將有限的審評資源聚焦在創(chuàng)新產(chǎn)業(yè)發(fā)展方向上��,監(jiān)管機構已在不斷優(yōu)化和調(diào)整醫(yī)療器械的風險分類。本文對比了中美兩國醫(yī)療器械分類結構的差異及分類動態(tài)調(diào)整的舉措�,為進一步完善我國醫(yī)療器械分類提供建議和思考��。
一、中美醫(yī)療器械分類構成差異明顯
統(tǒng)計美國《聯(lián)邦法規(guī)》第21條第862-892部分器械的分類情況�,F(xiàn)DA已將2000多種器械分成16個醫(yī)學領域�,Ⅰ類產(chǎn)品占比5%、Ⅱ類57%�、Ⅲ類38%�;我國是以技術領域為主線,從醫(yī)療器械功能和臨床使用的角度劃分產(chǎn)品歸屬�,包含22個子目錄�����,1700多種醫(yī)療器械,Ⅰ類產(chǎn)品占比26%��、Ⅱ類49%��、Ⅲ類25%�����。很明顯�,我國Ⅲ類產(chǎn)品占比明顯高于美國���,而Ⅰ類產(chǎn)品比例低于美國。某些類型的器械分類基本一致�����,比如消毒滅菌器械[1],而某些類型的器械分類差異比較大�����,比如心血管導管在我國主要歸為Ⅲ類�,在美國�,這類產(chǎn)品更多地被劃分為Ⅱ類���,Ⅲ類多為可能直接影響人體生命支持系統(tǒng)或可能導致嚴重損傷或疾病的醫(yī)療器械[2]��。
有研究表明���,F(xiàn)DA更側重于產(chǎn)品本身的危險性���,而NMPA更側重于管理的等級把控���,比如輸血輸液器具�,F(xiàn)DA認為該類產(chǎn)品本身的危險性并不高��,風險均可以通過特定手段控制��,因此歸為Ⅱ類��;而NMPA認為該類器械會直接侵入人體��,如果不嚴格管理則會對人體造成極大危害,于是歸為Ⅲ類[3]�����。此外����,美國更加強調(diào)企業(yè)自身的責任約束���,而我國更多是依靠NMPA的監(jiān)管和批準[4]。上述管理理念的差異一定程度上造成了中美兩國醫(yī)療器械風險分類的差異�。
理想的分類結構能夠?qū)徳u資源聚焦于真正高風險且具有臨床價值的創(chuàng)新產(chǎn)品上,進而加速產(chǎn)業(yè)升級����。由于醫(yī)療器械分類目錄是基于一定時期的監(jiān)管實踐和產(chǎn)業(yè)發(fā)展現(xiàn)狀制定的�����,隨著醫(yī)療器械行業(yè)的快速發(fā)展��,新技術���、新產(chǎn)品的不斷涌現(xiàn)��,在醫(yī)療器械產(chǎn)品的生產(chǎn)、經(jīng)營、使用過程中��,生產(chǎn)企業(yè)和監(jiān)管部門不斷積累經(jīng)驗�����,基于“新信息”對產(chǎn)品風險進行重新評估���,對管理類別進行動態(tài)調(diào)整尤為重要��。
二��、美國醫(yī)療器械分類調(diào)整的發(fā)展
美國是全球最早開始對醫(yī)療器械進行監(jiān)管的國家���,早在1938年��,《食品��、藥品和化妝品法案》(FD&C法案)就開始將監(jiān)管范圍擴大至醫(yī)療器械。面對醫(yī)療器械類型的繁多和技術的復雜�����,為監(jiān)管體系科學化、資源分配合理化���,1976年頒布了《醫(yī)療器械修正案》,該法案是一部全面的醫(yī)療器械法規(guī)���,引入了以產(chǎn)品風險為依據(jù)的三級分類系統(tǒng),提出了醫(yī)療器械上市前和上市后的管理制度,為新醫(yī)療器械進入市場建立了上市前批準(PMA)和上市前通知(510(k))的監(jiān)管途徑���,并在19世紀80年代中期完成初步分類��。
FDA一直以嚴格監(jiān)管聞名,廣泛贊譽的同時也面臨著各方的壓力,其中包括注冊程序繁雜����、審批時間過長等問題�。為此���,F(xiàn)DA在保障產(chǎn)品安全性的同時持續(xù)創(chuàng)新監(jiān)管舉措����,采取科學監(jiān)管手段提高審批效率��。其中����,對醫(yī)療器械分類的動態(tài)調(diào)整便是一項重要的舉措�,以促進和加快醫(yī)療器械的研發(fā)和審查。美國作為醫(yī)療器械監(jiān)管的先行者�����,其管理類別動態(tài)調(diào)整機制從1976年建立至今�,通過動態(tài)調(diào)整維持了醫(yī)療器械分類系統(tǒng)的穩(wěn)定有效運行�����。
·FDA對醫(yī)療器械的分類實行動態(tài)調(diào)整���,踐行“最小負擔原則”的管理理念��。
根據(jù)FD&C法案��,分類調(diào)整可由FDA自行啟動或回應利益相關者的重新分類申請��。在進行分類調(diào)整時,F(xiàn)DA會結合醫(yī)療器械分類小組的意見����,確定擬定分類及相關證據(jù)并在《聯(lián)邦公報》上公示,結合公眾意見進一步審查考慮����,隨后發(fā)布最終的分類、證據(jù)及對公眾意見的解釋���。當產(chǎn)品風險充分識別,并且能夠通過針對性控制措施避免風險���,則產(chǎn)品分類降級���;當產(chǎn)品上市后臨床研究以及不良事件數(shù)據(jù)顯示安全性存在各類風險�,有效性不足,正面風險-收益特征未能建立�,現(xiàn)有管控措施不能建立起針對預期用途的安全性和有效性的合理保證��,則產(chǎn)品分類升級[5]����。
2012年����,國會頒布了《食品和藥品管理局安全與創(chuàng)新法》(FDASIA)���,推動藥品和醫(yī)療器械審評體制的改革��,該法案中要求FDA每年公布前一年重新分類的情況��。為促進和加快高風險醫(yī)療器械的發(fā)展和審核���,2014-2015年FDA完成了戰(zhàn)略重點項目——對PMA醫(yī)療器械的回顧性審核。該項目分三個階段對通過PMA上市的產(chǎn)品結合臨床研究����、非臨床數(shù)據(jù)以及實際臨床表現(xiàn)等信息進行分析���,在保障產(chǎn)品安全的前提下,考慮上市前/后數(shù)據(jù)收集要求��,判斷是否可以降低器械的分類�����,進而加快產(chǎn)品的上市進程�、簡化上市流程[6]���。
值得注意的是���,F(xiàn)DA對醫(yī)療器械的分類調(diào)整并非僅關注高風險產(chǎn)品的降級�����。經(jīng)統(tǒng)計�,2013-2021年�,共有22個產(chǎn)品由Ⅲ類調(diào)整為Ⅱ類,1個產(chǎn)品由Ⅱ類調(diào)整為Ⅲ類����,5個產(chǎn)品由Ⅰ類調(diào)整為Ⅱ類[7]�。做出分類調(diào)整的判斷是基于產(chǎn)品“新信息”的分析研究,這些“新信息”則在于全生命周期數(shù)據(jù)系統(tǒng)的支持以及嚴格的上市后監(jiān)管信息的補充�。
·對醫(yī)療器械進行分類調(diào)整��,需要前端信息集合的支持、后端監(jiān)管的保障以及專家的支持�。
FDA對醫(yī)療器械實施的層次化�、強制性的上市后監(jiān)管是分類結構不斷優(yōu)化的堅實保障。一是通過不良事件上報制度以及安全監(jiān)測網(wǎng)絡�����,F(xiàn)DA常態(tài)化對醫(yī)療器械的安全性進行監(jiān)管��、識別安全性問題��;二是開展預防性監(jiān)測,部分Ⅱ類���、Ⅲ類醫(yī)療器械需要生產(chǎn)企業(yè)制定研究計劃并得到FDA的審批���,主動�����、系統(tǒng)��、科學��、有效地收集已上市醫(yī)療器械的數(shù)據(jù)��,并對數(shù)據(jù)及其他信息進行分析和判斷,預測不良事件的實際發(fā)生率��,提交上市后監(jiān)管的中期報告以及最終報告��;三是定期的質(zhì)量體系的監(jiān)管核查���。FDA對Ⅰ類產(chǎn)品每四年檢查一次質(zhì)量體系,對Ⅱ��、Ⅲ類產(chǎn)品每兩年檢查一次質(zhì)量體系[8]�。
FDA有強大的�、全面的醫(yī)療器械數(shù)據(jù)系統(tǒng)���,能夠資源集成���、累積經(jīng)驗。FDA充分利用信息化手段進行監(jiān)管��,2021年����,F(xiàn)DA提出數(shù)字化轉(zhuǎn)型倡議���,以改善內(nèi)部和外部客戶體驗�����、提高申請材料的質(zhì)量和審查效率[9]。FDA建立的數(shù)據(jù)庫組相互獨立又有機聯(lián)系���,覆蓋了監(jiān)管法規(guī)���、產(chǎn)品分類���、注冊�、上市后監(jiān)管整個監(jiān)管鏈條[10]�����。醫(yī)療器械分類動態(tài)調(diào)整的信息基礎就是上市前后臨床研究、臨床數(shù)據(jù)��、技術發(fā)展等帶來的新信息�����、新經(jīng)驗�����,通過對數(shù)據(jù)庫信息的挖掘與分析�,做出科學可靠的判斷���。
FDA有充足的審評專員、專家資源的支持�����。2023年,CDRH有2230名專職人員[11]�����;此外�,F(xiàn)DA設立了包括18個小組的醫(yī)療器械咨詢委員會��,每個小組由一位該領域的醫(yī)學專家和六位學者組成���,成員由臨床、工程�����、生物和物理科學以及其他相關的專業(yè)中選出�,其中包括無表決權的消費者和行業(yè)代表���。這18個小組每年定期召開會議,對已經(jīng)上市或正接受檢查的產(chǎn)品查看和評估其安全性與有效性的相關資料���,除醫(yī)療器械爭議解決小組外��,每個小組對專業(yè)領域內(nèi)的醫(yī)療器械產(chǎn)品的安全性和有效性的相關事宜給出建議�,包括討論醫(yī)療器械產(chǎn)品分類和重新分類的相關問題�����。
三、中國醫(yī)療器械分類制度改革及思考
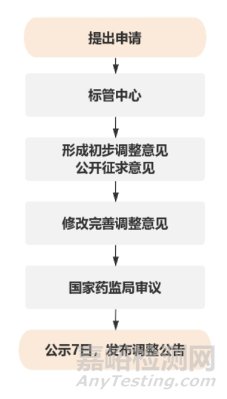
2000年�,我國發(fā)布《醫(yī)療器械監(jiān)督管理條例》(以下簡稱《條例》)標志著對醫(yī)療器械開始實施更為規(guī)范的監(jiān)管[12]����。隨后,2002年發(fā)布了《醫(yī)療器械分類目錄》(以下簡稱《分類目錄》)���,該《分類目錄》是對1998年版的全面修訂���。為適應監(jiān)管和產(chǎn)業(yè)發(fā)展需要����,2017年原國家食品藥品監(jiān)督管理局發(fā)布了新修訂的《分類目錄》��,成立醫(yī)療器械分類技術委員會作為分類管理工作的技術支撐。2021年�����,《條例》再次修訂發(fā)布�����,以法規(guī)形式鞏固制度改革和“放管服”改革成果�;同年��,落實《條例》對分類目錄進行調(diào)整的規(guī)定����,發(fā)布《醫(yī)療器械分類目錄動態(tài)調(diào)整工作程序》��,重視各利益相關方的建議和意見���,見圖1���。其實在2016年���,原國家食藥監(jiān)局就開始討論醫(yī)療器械分類目錄動態(tài)調(diào)整機制的建設[13]����。
圖1 利益相關方申請分類調(diào)整的流程備注:注冊人和備案人����、生產(chǎn)經(jīng)營企業(yè)、使用單位提出的申請應先經(jīng)過省級藥監(jiān)部門的初審�,必要的由省級藥監(jiān)部門提出申請。
我國監(jiān)管部門順應科學規(guī)律����、適應產(chǎn)業(yè)創(chuàng)新發(fā)展�、貼近臨床應用需求,持續(xù)優(yōu)化醫(yī)療器械的分類�����。2017年版《分類目錄》發(fā)布后�,我國共發(fā)布了四次調(diào)整《醫(yī)療器械分類目錄》部分內(nèi)容的公告���,分類調(diào)整的范圍并未僅限于高風險產(chǎn)品�����。經(jīng)統(tǒng)計����,有18種產(chǎn)品由Ⅲ類降為Ⅱ類����,11種產(chǎn)品由Ⅱ類降為Ⅰ類,4種產(chǎn)品由Ⅰ類升為Ⅱ類��,1種產(chǎn)品由Ⅱ類升為Ⅲ類���;此外,還有3種產(chǎn)品細化產(chǎn)品描述或預期用途之后降低風險分類��,比如眼科激光診斷設備���,如果發(fā)生強激光時保持Ⅲ類,如果發(fā)生弱激光則歸為Ⅱ類����,有1種產(chǎn)品細化預期用途后升級風險分類���,超聲手術設備用于骨組織時保持Ⅱ類�,用于手術中對血管、軟組織及器官進行切割�����、止血和血管閉合時歸為Ⅲ類��。
醫(yī)療器械具有多樣化、專業(yè)領域跨度大等特點�����,對其進行科學分類是有效監(jiān)管�����、合理配置監(jiān)管資源的重要基礎[14]。醫(yī)療器械的分類調(diào)整是全面���、持續(xù)且循序漸進的過程�����,是基于信息的不斷收集與分析做出的科學判斷�����。為進一步促進分類管理適應產(chǎn)業(yè)的發(fā)展����,以“放管服”為原則[15]�,還應重視以下問題:
一是加強審評隊伍人力和能力建設�,緊跟產(chǎn)業(yè)發(fā)展����。審評人員的經(jīng)驗積累是分類優(yōu)化的基礎支撐,不僅能根據(jù)審評經(jīng)驗提出分類調(diào)整建議�����,并且為風險調(diào)整后的管理提供依據(jù)�。CMDE公示的審評人員不足200人,另外由于區(qū)域間經(jīng)濟水平差異較大�����、行業(yè)發(fā)展的不平衡�����,不同省份審評隊伍能力也有一定的差異[16]�,但創(chuàng)新醫(yī)療器械的快速發(fā)展對審評人員的專業(yè)水平有著極高的要求�����。打造高水平的審評隊伍��,應加強政企間交流學習,緊跟技術發(fā)展速度���,適時調(diào)整產(chǎn)品分類,提高審評效率�。
二是上市后監(jiān)管細化落實,強化企業(yè)責任意識��,提高上市后信息可利用度�。目前,我國上市后信息的有效性不足���,比如不良事件監(jiān)測系統(tǒng)內(nèi)數(shù)據(jù)來自企業(yè)報告比例小���,報告質(zhì)量有待提高��,追溯制度不完整等[8]����。創(chuàng)新醫(yī)療器械在審評時風險并不能充分識別���,因此應強化上市后的監(jiān)管���。利用信息系統(tǒng)補充對產(chǎn)品風險的認識之外,還應加強企業(yè)自身責任意識�����,完善創(chuàng)新器械上市后的報告制度��,通過上市后信息的反饋輔助研判產(chǎn)品風險分類的合理性以及后續(xù)產(chǎn)品的上市前審評��。
三是基礎設施建設提高工作效率���。我國雖建立了醫(yī)療器械注冊��、標準�、分類、唯一標識����、不良反應等多個數(shù)據(jù)庫��,但各個數(shù)據(jù)庫相互獨立[8]���。比如在面對有些產(chǎn)品由Ⅲ類調(diào)整為Ⅱ類時,產(chǎn)品批準部門改變���、申報系統(tǒng)不同,審評人員無法直接了解歷往的注冊信息���,由此可能造成一些不必要的分類法補��,損失效率[17]����。充分利用信息化監(jiān)管模式,尤其是系統(tǒng)間的銜接�,幫助全國監(jiān)管部門實現(xiàn)信息共享[18],打造“一盤棋”審評模式���,統(tǒng)一審評標準,提升監(jiān)管效率��。同時運用大數(shù)據(jù)���、人工智能等技術不斷收集、分析產(chǎn)品上市前后的信息�,完善對產(chǎn)品風險的認知,也有助于產(chǎn)品分類調(diào)整及監(jiān)管���。
綜上�����,醫(yī)療器械的分類是制度改革的基礎��,關乎著資源的合理配置與審評制度改革系統(tǒng)工程的運轉(zhuǎn)����;反過來�����,制度的持續(xù)優(yōu)化也會推動分類的調(diào)整��,兩者互相推動�����。在未來還應考慮���,分類的不斷調(diào)整�����,Ⅱ類產(chǎn)品隨之增多���,省級審評與監(jiān)管能力如何適應����。我國醫(yī)療器械監(jiān)管制度改革起步較晚���,經(jīng)驗不足,故而最初的分類相對保守也屬情理之中����,但隨著監(jiān)管核心問題的深入探討��,改革進程會不斷加速�����。
參考文獻:
1.譚瑞芬,胡昌明,張春青.醫(yī)療器械消毒滅菌器械國內(nèi)外分類管理探討[J].中國藥事,2018,32(09):1176-1180.
2.美國醫(yī)療器械的分類鑒定. https://zhuanlan.zhihu.com/p/676169331
3.思宇醫(yī)械觀察.一文看懂���!中美醫(yī)療器械注冊有什么區(qū)別�����?https://www.innomd.org/article/5efd3f27218ba117925170b6
4.飛速度醫(yī)療器械咨詢. 泛談中美醫(yī)療器械的分類管理. https://flyingspd.com/news/basics/5919.html
5.張春青, 周良彬, 王越等. 美國醫(yī)療器械管理類別動態(tài)調(diào)整機制及其工作思路探究[J]. 中國醫(yī)療器械雜志, 2021, 45 (03): 315-320.
6.Medtec. 淺談美國FDA 高風險醫(yī)療器械審批要求的最新調(diào)整思路. https://www.medtecchina.com/regulation/4790/
7. FDA. Reclassification. https://www.fda.gov/about-fda/cdrh-transparency/reclassification
8.郭艷,慶凌,關懷.中美醫(yī)療器械上市后監(jiān)管模式對比[J].中國藥業(yè),2023,32(06):4-7.
9.曾美琪,連小奇.美國FDA醫(yī)療器械智慧監(jiān)管現(xiàn)狀與啟示[J].中國醫(yī)藥導刊,2023,25(06):565-569.
10.王越,周良彬,王悅,李健,張春青.美國和日本醫(yī)療器械數(shù)據(jù)庫系統(tǒng)構建思路探析[J].中國藥事,2019,33(10):1181-1186.
11.CDRH.2023 Annual report.
12.譚瑞芬,胡昌明,張春青.醫(yī)療器械消毒滅菌器械國內(nèi)外分類管理探討[J].中國藥事,2018,32(09):1176-1180.
13.國家藥監(jiān)局. 醫(yī)療器械分類目錄探討會在京召開. https://www.nmpa.gov.cn/ylqx/ylqxjgdt/20161122172401675.html
14.中國醫(yī)藥報. 制度創(chuàng)新助推行業(yè)高質(zhì)量發(fā)展分類目錄動態(tài)調(diào)整賦能精準化管理. https://www.samr.gov.cn/xw/df/art/2023/art_a838aac1f0f5421ba66dac2926b25260.html
15.淺談監(jiān)管科學:評估、框架與創(chuàng)新. https://new.qq.com/rain/a/20200831A0QSH900
16.全國體外診斷網(wǎng) CAIVD. 我國醫(yī)療器械注冊管理制度存在的問題及建議. https://mp.weixin.qq.com/s/BXuZRgfVlTFlv3vhKZeNeA
17.楊雨萌,張威鵬,符夢凡.淺談新版《醫(yī)療器械分類目錄》實施后涉及管理類別調(diào)整的第二類敷料類產(chǎn)品在延續(xù)注冊過程中的常見問題[J].中國醫(yī)療器械信息,2023,29(01):7-10.
18.向麗烜,蘇紫媚,張梓鋒等.我國醫(yī)療器械監(jiān)管問題解析及其優(yōu)化策略探討[J].中國醫(yī)療器械信息,2023,29(07):8-11