一��、概述
自2019年12月1日MAH制度在中國正式施行以來���,藥品共線生產(chǎn)的普遍性和藥品質(zhì)量安全監(jiān)管的緊迫性成為藥企和藥監(jiān)部門亟需面對和重視的問題。為了規(guī)范和指導藥品共線生產(chǎn)管理,避免產(chǎn)品間的污染和交叉污染�����,NMPA在2021年11月發(fā)布了《藥品共線生產(chǎn)質(zhì)量管理指南(征求意見稿)》�����,并于2023年03月正式頒布了《藥品共線生產(chǎn)質(zhì)量風險管理指南》。指南的頒布不僅為國內(nèi)藥品生產(chǎn)企業(yè)的共線生產(chǎn)管理和風險評估提供了法規(guī)指導��,而且為企業(yè)提供了相對系統(tǒng)的評估策略�����、方法和標準。在本文中�����,本人結合自己的經(jīng)驗和理解��,重點剖析和闡述了上述指南中關于藥品共線生產(chǎn)的基本原則和要求,以期共同提升對該問題的認識��,同時也歡迎同仁補充和指正��。
二�����、藥品共線生產(chǎn)的基本原則
根據(jù)《藥品共線生產(chǎn)質(zhì)量風險管理指南》的定義��,藥品共線生產(chǎn)是指多種藥品共用生產(chǎn)線進行生產(chǎn)�����,包括共用生產(chǎn)廠房���、設施和設備,但不包括共用質(zhì)量控制實驗室���、庫房���、取樣間等輔助設施和儀器。指南中關于藥品共線生產(chǎn)的基本原則共計5條���,見表1�����。
表1 藥品共線生產(chǎn)的基本原則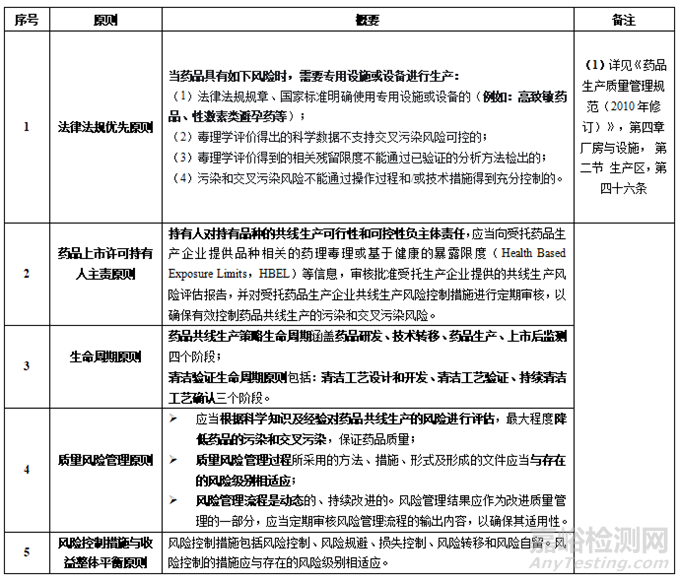
三��、藥品共線生產(chǎn)評估
藥品共線生產(chǎn)范圍涵蓋商業(yè)化產(chǎn)品���、非商業(yè)化規(guī)模的試制樣�����、臨床試驗藥品等���,涉及共線情況比較多樣,例如:①臨床試驗用藥品與商業(yè)化藥品共線���;②中藥產(chǎn)品共線�����;③生物制品共線��;④最終滅菌產(chǎn)品與非最終滅菌產(chǎn)品共線���;⑤某些激素類��、細胞毒性類��、高活性化學藥品共線��;⑥基因治療與細胞治療產(chǎn)品共線�����;⑦麻醉藥品���、精神藥品和藥品類易制毒化學品共線;⑧青霉素類及β-內(nèi)酰胺結構類產(chǎn)品共線等���。以上哪些情況可以共線生產(chǎn)��?哪些不可以就需要具體分析和評估。
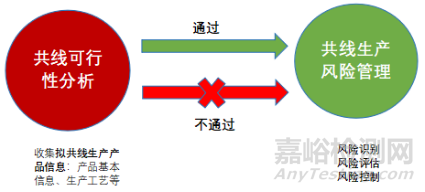
共線生產(chǎn)評估包括兩部分���,共線可行性分析和共線生產(chǎn)風險管理���,其基本邏輯關系見圖1。
圖1 共線可行性分析和共線生產(chǎn)風險管理
共線生產(chǎn)風險管理(風險識別���、評估和控制)是消除或降低共線生產(chǎn)風險并確保藥品質(zhì)量和安全的關鍵環(huán)節(jié)�����,共線生產(chǎn)企業(yè)必須嚴格執(zhí)行和強化管理�����。
3.1共線可行性分析
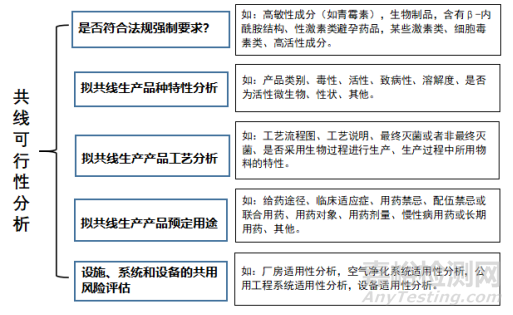
在實施多產(chǎn)品共線生產(chǎn)時���,我們必須意識到完全消除產(chǎn)品間的交叉污染風險是不現(xiàn)實的��。在GMP條件下��,需要將交叉污染的控制程度與確定的患者風險水平相匹配�����。生產(chǎn)企業(yè)應遵守國家法規(guī)條例的基礎上�����,根據(jù)企業(yè)質(zhì)量體系特點�����,結合科學知識及經(jīng)驗實施風險管理�����。共線可行性分析是第一步���,相關內(nèi)容請參見圖2��。
圖2 共線可行性分析
如果通過分析發(fā)現(xiàn)不可以共線生產(chǎn)(參見 表1藥品共線生產(chǎn)的基本原則中相關描述)���,則終止共線或改為專線生產(chǎn)。如果通過共線可行性分析��,可以共線生產(chǎn)���,則啟動共線生產(chǎn)風險管理-風險評估(識別���、分析、評價)和控制�����,進而消除或降低共線生產(chǎn)風險�����,最終保障藥品的質(zhì)量和安全��。
3.2 共線生產(chǎn)風險管理
藥品共線生產(chǎn)風險管理的目的是確保在共線生產(chǎn)過程中不同藥品之間不會相互污染或影響產(chǎn)品質(zhì)量���。共線生產(chǎn)風險管理的關鍵措施如下:
風險識別:確定潛在風險��,包括交叉污染��、設備故障��、人為錯誤等�����。
風險評估:評估各種潛在風險的可能性和影響程度��,以確定應對重點�����。
控制措施:制定控制措施�����,如采取物理隔離��、清潔程序�����、不同工藝區(qū)域間的限制等���,以防止交叉污染�����。
有關共線生產(chǎn)評估和控制的基本要求��,請參見表2��。
表2 共線生產(chǎn)評估和控制基本要求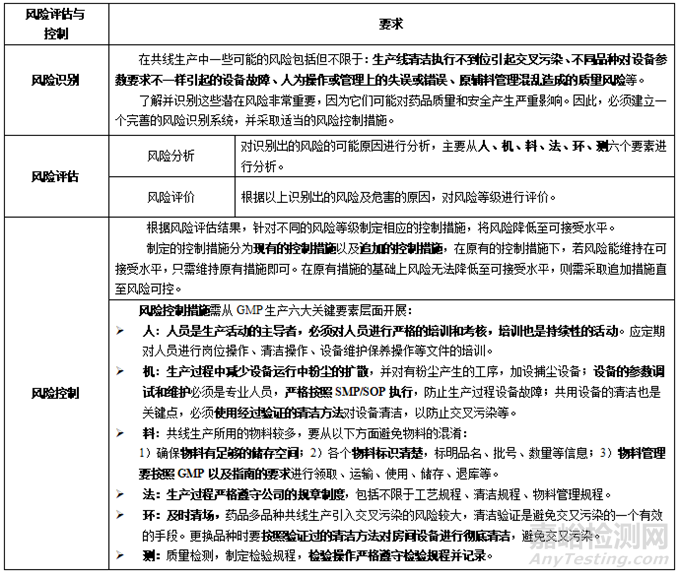
3.3 案例分析
背景:以某公司XX生產(chǎn)車間擬共線A��、B品種小規(guī)格最終滅菌注射劑為例�����。
流程:藥品共線生產(chǎn)評估致流程包括:1)組建評估小組��;2)收集產(chǎn)品信息�����;3)共線可行性分析���;4)風險識別;5)風險評估���;6)風險控制���;7)風險回顧。
1)組建評估小組
建立一個評估小組���,使相關領域有經(jīng)驗有技術的人員參與到風險評估中來���,最大限度抵消主觀行為偏見的影響。公司選擇的評估團隊���,一般來自于各部門(車間��、質(zhì)量��、設備部�����、研發(fā)���、倉儲)部長�����、主管或技術骨干�����,他們經(jīng)驗豐富�����,在自己的領域小有成就并能獨當一面��。
2)收集產(chǎn)品信息
共線生產(chǎn)的注射劑產(chǎn)品應有相似或相近的產(chǎn)品特性��,應對具體的產(chǎn)品特性進行分析�����,包括毒性���、致敏性���、活性�����、溶解度�����、性狀和配伍禁忌或相互反應等��,以確定多品種共線生產(chǎn)的可行性�����。
3)共線可行性分析
① 法規(guī)要求
根據(jù)《藥品共線生產(chǎn)質(zhì)量風險管理指南》(2023年3月)和《藥品生產(chǎn)質(zhì)量管理規(guī)范》(2010年修訂)相關規(guī)定�����,評估共線產(chǎn)品是否滿足相關法規(guī)要求��,見表3。
表3 法規(guī)要求符合性分析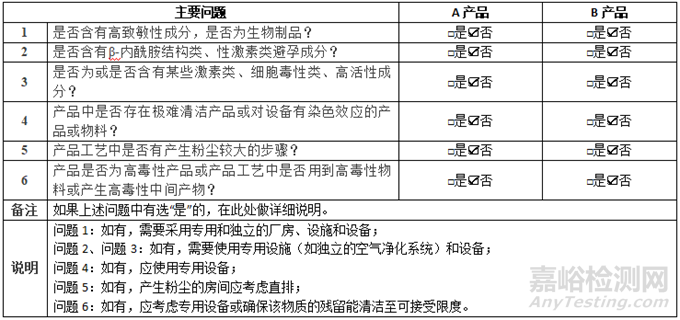
②廠房���、設施和設備的適用性
表 4 廠房��、設施和設備的適用性分析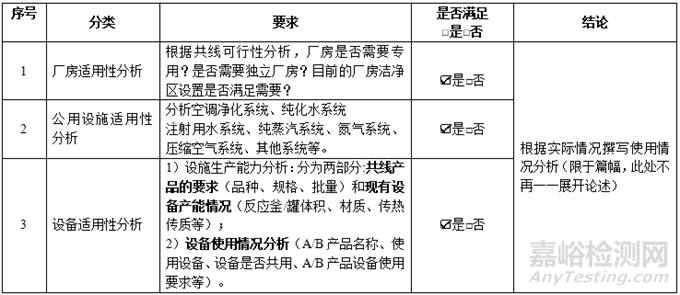
③ 工藝可行性分析
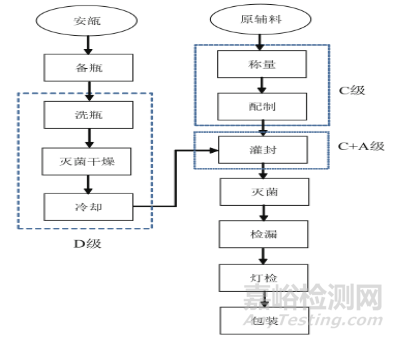
最終滅菌小容量注射劑生產(chǎn)為例���,其主要工序包括如圖3所示。
圖3 最終滅菌小容量注射劑一般工藝流程
通過工藝流程分析��,需要考慮每道工序的生產(chǎn)過程中可能發(fā)生混淆和交叉污染的風險點�����?����?紤]到人���、機��、料�����、法��、環(huán)��、測等六個方面�����,以確保不遺漏任何風險點�����。重點分析關鍵生產(chǎn)工藝是否容易受共線生產(chǎn)的另一產(chǎn)品的干擾��,而最終滅菌小容量注射劑的關鍵生產(chǎn)工藝包括配制�����、灌封和滅菌��。
4)風險識別
多產(chǎn)品共線生產(chǎn)中產(chǎn)生危害的主要原因�����,可以從人�����、機���、料���、法、環(huán)��、測幾個角度來分析���,常見的原因與示例�����,見表 5���。
表5 多產(chǎn)品共線生產(chǎn)主要風險常見原因分析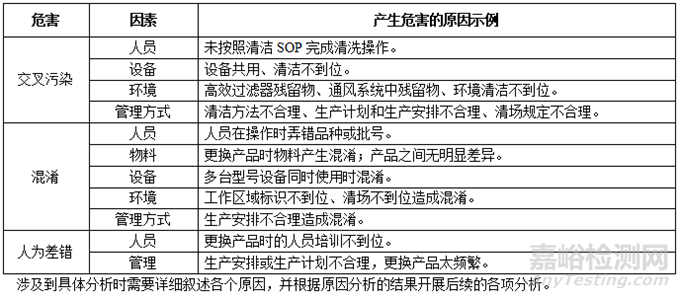
5)風險評估
① 風險等級評價表
評價每項風險要素的失效影響,用S(嚴重度)�����、O(可能性)�����、D(可探測度)三個指標,每個指標劃分5級��,對應分數(shù)均為1~5分���,每級設定評價標準(限于篇幅��,此處省略分級和評價標準的具體內(nèi)容)��。最后計算風險系數(shù)RPN=S*D*P��。RPN值在1~125之間���,根據(jù)公司抗風險能力及公司實際情況��,將風險水平等級設定為低風險�����、中等風險���、高風險��,見表6���。
表6 風險等級評價表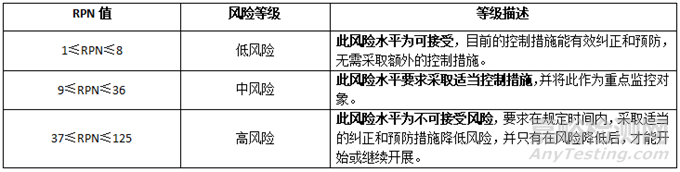
② 風險評估表
表7 A�����、B品種最終滅菌小容量注射劑風險評估表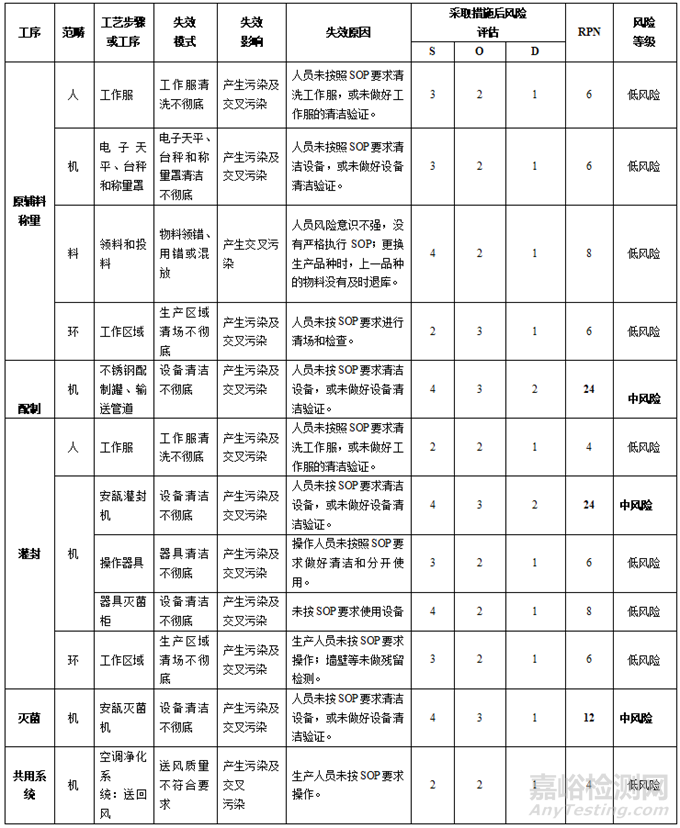
6)風險控制
針對表7中及以上風險的各風險要素需制定針對性控制措施�����,并重新進行風險評估���,直至風險水平降至可接受水平(低風險),見表8�����。
表 8 最終滅菌小容量注射劑A���、B品種共線生產(chǎn)采取措施后的風險評估結果表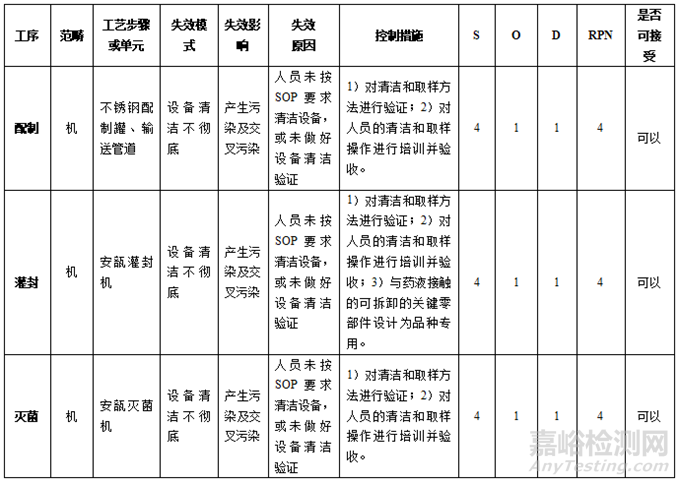
經(jīng)過風險評估��,要降低XX車間最終滅菌小容量注射劑A�����、B品種共線生產(chǎn)的風險��,必須確保涉及到的各項風險控制措施都得到落實�����,并且能夠達到預期效果后才能放行所生產(chǎn)的產(chǎn)品��。
7)風險回顧
完成風險評估后���,應定期回顧風險管理過程的結果�����,回顧頻率應與相應的風險水平相適應��。建立定期的風險審查機制��,分析產(chǎn)品各項指標控制情況,總結偏差特點和趨勢���,并制定風險降低改進計劃���。
此外�����,企業(yè)還應在以下情況下重新評價風險:①當原輔料��、直接接觸藥品的包裝材料�����、質(zhì)量標準��、檢驗方法��、操作規(guī)程���、廠房、設施��、設備���、儀器��、生產(chǎn)工藝��、計算機軟件等發(fā)生變更時��;②當法律法規(guī)及技術要求發(fā)生變更時�����;③在現(xiàn)有生產(chǎn)線增加品種時�����;④當企業(yè)的管理層或客戶提出對質(zhì)量管理更高的要求時���。
8)結論
通過對某公司XX車間的最終滅菌小容量注射劑A��、B的共線可行性分析(法規(guī)要求���、廠房、設施和設備�����、工藝等)���,經(jīng)過風險識別和評估,發(fā)現(xiàn)客觀上存在交叉污染、混淆和人為差錯的風險���,針對中高風險要素通過風險控制措施改進后���,這些要素的風險等級降低至可接受水平,故最終認為XX車間針對上述品種共線生產(chǎn)是可行的��,共線生產(chǎn)期間需要對控制措施進行有效的維護�����,定期對風險進行回顧��。
四�����、結束語
藥品生產(chǎn)企業(yè)多品種共線能夠整合資源��、降低成本��、提高效率���。自《藥品管理法》及“藥品上市許可持有人制度”執(zhí)行后���,產(chǎn)生了大量的MAH和CMO/CDMO企業(yè)���,持有人與藥品生產(chǎn)的分離,對藥品共線生產(chǎn)的需求也在增加�����。藥品是特殊的商品���,藥品的質(zhì)量和安全直接影響患者的生命健康,故藥品生產(chǎn)企業(yè)在實施藥品共線生產(chǎn)時必須嚴格遵守國家的藥政法規(guī)規(guī)定�����,結合企業(yè)自身特點��,運用科學知識和經(jīng)驗���,做好藥品共線生產(chǎn)風險管理。首要是進行共線生產(chǎn)可行性評估�����,經(jīng)過評估可以共線生產(chǎn)的前提下,需進一步做好風險管理(風險識別���、評估、控制)�����,制定適合企業(yè)的SMP和SOP制度規(guī)范��,做好人員培訓工作���,完善清潔驗證等質(zhì)量管理工作��,最終確保共線生產(chǎn)過程的高效和合規(guī)�����。
參考文獻
[1] 《藥品共線生產(chǎn)質(zhì)量風險管理指南》國家藥品監(jiān)督管理局,2023-03-06.
[2] 《藥品生產(chǎn)質(zhì)量管理規(guī)范》衛(wèi)生部令第79號���,2011-01.01.
[3] 《中華人民共和國藥品管理法藥品管理法》.國家市場監(jiān)督管理總局��,2019-08-26.
[4] 樓雙鳳, 曹輝, 張闖. 藥品共線生產(chǎn)問題分析及建議[J].中國醫(yī)藥工業(yè)雜志, 2022, 53(7):4.
[5] 田文淼,梁毅.小容量注射劑共線生產(chǎn)的質(zhì)量風險控制研究[J].中國醫(yī)藥工業(yè)雜志, 2020, 51(6):6.