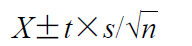摘要
外用藥物制劑具有復雜的藥物遞送途徑,通常局部起效�����,輔料成分復雜����,處方組成和制備工藝參數(shù)的微小差異就有可能導致產(chǎn)品不同的質量特性,從而影響藥物的安全性和有效性��。體外釋放試驗(IVRT)可用于表征一個產(chǎn)品批次的穩(wěn)態(tài)藥物釋放速率��,表征某些工藝����、配方和/或生產(chǎn)的變更對藥品的影響,在某些情況下�����,可用于論證產(chǎn)品批量放大或上市后變更的等效性��。盡管IVRT不能模擬體內(nèi)性能情況��,但其仍是一項關鍵質量屬性��,應訂入產(chǎn)品放行和貨架期質量標準����。本文參考國內(nèi)外相關技術指導原則及文獻����,對皮膚外用制劑的IVRT研究技術要求概況進行綜述�����,期望為業(yè)內(nèi)人士提供參考����。
關鍵詞
外用藥物制劑;經(jīng)皮給藥��;體外釋放試驗����;一致性評價
外用藥物制劑相較于口服、注射劑等給藥方式具有許多優(yōu)勢:①避免肝臟的首過效應����;②藥物吸收不受胃腸道內(nèi)pH值、食物及其他藥物等因素的影響����;③具有控釋效果��;④維持恒定的有效血藥濃度����,避免因吸收過快產(chǎn)生血藥濃度過高引起不良反應��;⑤給藥方便��,安全性高,患者順應性好[1]�����。已成為現(xiàn)代醫(yī)學中重要的組成部分����,同時具有較好的發(fā)展前景。
外用藥物制劑是一類發(fā)揮局部或全身治療作用的制劑����,劑型包括軟膏劑、乳膏劑��、凝膠劑����、散劑�����、乳劑�����、糊劑��、混懸劑��、噴霧劑�����、氣霧劑�����、溶液和其他半固體和/或液體劑型[2-3]�����。其中����,乳膏劑、軟膏劑及凝膠劑處方組成復雜����,多為半固體制劑,涉及油相�����、水相及油包水/水包油等熱力學不穩(wěn)定體系��,使用多種不同性質的輔料(油相����、水相��、表面活性劑��、矯味劑��、清涼劑����、抑菌劑等)��。相對于傳統(tǒng)的注射劑和片劑等劑型��,其制備和穩(wěn)定性過程中的動力學和熱力學影響因素眾多�����,在設計��、制造�����、儲存和運輸?shù)榷鄠€環(huán)節(jié)均存在較大的挑戰(zhàn)����。
對于該類制劑�����,藥物在體外釋放的速率和程度是其性能的綜合體現(xiàn)����,主要用于產(chǎn)品的藥學質量控制,也可用于藥品開發(fā)過程中處方工藝的篩選研究��。雖然不能根據(jù)體外釋放試驗(IVRT)釋放速率的差異預測體內(nèi)生物利用度的變化[4],但其具有潛在反應制劑中藥物生物學特性變化的能力�����,而這種特性的變化可能與以下因素有關����,如處方中的活性或非活性組分、制劑的物理化學特性�����、生產(chǎn)工藝的變更����、運輸和儲存條件��、產(chǎn)品的放置時間長短和其他對終產(chǎn)品質量特性具有關鍵作用的因素等[5]��。
基于制劑的定性(Q1)��、定量(Q2)以及物理化學和結構特性(Q3)�����,Shah等[6]于2015年提出了外用藥物分類系統(tǒng)(TCS),該系統(tǒng)將皮膚外用制劑分為4類����。如果仿制藥和參比制劑(RLD)之間的Q1、Q2和Q3都相同�����,則仿制藥可能實現(xiàn)生物等效性豁免(TCS1類)��。在TCS理念中����,某些輔料可能在藥物傳遞、發(fā)揮作用和隨之產(chǎn)生的療效中起到積極作用����。但總體來看,許多輔料是惰性的��。如果仿制藥和RLD之間Q1��、Q2不同��,但在充分評估輔料的安全性和功效后,可進行IVRT評價����,若仿制藥和RLD一致,則可以實現(xiàn)生物等效性豁免(TCS3類)��。
歐洲藥品管理局(EMA)允許通過證明Q3及IVRT的等效性�����,用于支持單相的溶液劑�����、凝膠劑����、軟膏劑等簡單的配方制劑與參比制劑相比的治療等效性,以代替等效性臨床試驗[7]��。美國食品藥品監(jiān)督管理局(FDA)要求����,對于外用半固體制劑����,如果仿制藥的Q3與對照標準制劑相同��,僅需提供有限的其他證據(jù)(體外����、in silico和/或體內(nèi))�����,即可支持生物等效性論證[8]����,如根據(jù)2022年10月之后FDA更新的特定藥品指南,在Q1�����、Q2和Q3一致的基礎上��,阿達帕林凝膠[9]����、乳酸銨乳膏[10]、鹽酸布替萘芬乳膏[11]����、磷酸克林霉素凝膠[12]和酒石酸溴莫尼定凝膠[13]等僅需要論證IVRT等效��,即可豁免臨床等效性研究�����。日本醫(yī)藥食品局(PMDA)可通過IVRT研究對已上市半固體制劑和貼劑的B水平處方變更進行論證[14-15]��。
綜上可知����,IVRT研究在外用藥物制劑處方開發(fā)����、生物等效性論證以及上市后變更中的重要性,因此對IVRT研究進行合理設計�����,確保其結果準確可靠十分重要�����。雖然FDA����、EMA和PMDA均有發(fā)布關于IVRT研究的相關指南,但目前國內(nèi)暫無統(tǒng)一的技術要求規(guī)范����,且各國技術要求亦不統(tǒng)一,因此本文對IVRT研究相關技術要求概況進行總結�����,期望為業(yè)內(nèi)人士提供參考��。
1��、國內(nèi)外政策法規(guī)
在2018年國家藥品監(jiān)督管理局藥品審評中心(CDE)發(fā)布的《新注冊分類的皮膚外用仿制藥的技術評價要求(征求意見稿)》中闡述可參照FDA SUPAC-SS指南[16]進行IVRT研究[17]����,但在2021年發(fā)布的《皮膚外用化學仿制藥研究技術指導原則(試行)》中,建議參考FDA��、EMA和PMDA相關指導原則開展相關研究工作�����。
在1997年FDA發(fā)布SUPAC-SS指南中首次對IVRT研究的相關參數(shù)及數(shù)據(jù)統(tǒng)計進行了闡述�����,包括上樣量(300mg)、上樣方式(閉環(huán))�����、接收介質(水性緩沖液或水醇混合溶液)�����、合成膜����、試驗時長和取樣點(至少5個取樣點)等。2016年發(fā)布的《阿昔洛韋指南草案》[18]是第一份關于IVRT方法驗證及接受標準的指南�����,但其部分參數(shù)(如線性與范圍����、專屬性)的接受標準相對2022年10月FDA發(fā)布IVRT指南[3]較為寬松,目前已被FDA IVRT指南替代��。2022年5月美國藥典(USP)發(fā)布的《PF48(3)<1724>Semisolid drug products-performance tests》(以下簡稱USP<1724>)是現(xiàn)有國內(nèi)外藥典或指南中對IVRT的研究設計最為全面的法規(guī)指南[4]����,而FDA IVRT指南是專門闡述IVRT方法驗證的相關指南。
2010年PMDA發(fā)布的《皮膚局部適用制劑(半固體制劑和貼劑)處方變更生物等效性研究指南》中對IVRT的研究設計和數(shù)據(jù)統(tǒng)計進行了闡述�����,但未說明IVRT各參數(shù)的可接受標準�����,亦未對方法驗證內(nèi)容進行說明[14]�����。
2018年EMA發(fā)布的《外用制劑質量和等效性指南草案》中亦對IVRT研究設計的相關參數(shù)進行了闡述�����,但未明確膜惰性的可接受標準����、接收介質的組成,且在方法驗證模塊僅要求對區(qū)分力����、精密度和耐用性進行評估[7]�����。
USP����、FDA��、EMA和PMDA對IVRT研究設計的技術要點如表1所示��。
2��、研究設計
2.1 設備
在IVRT研究中��,可用的設備包括立式擴散池(VDC)�����、浸沒池�����、USP裝置4(流通池)����、USP裝置5(漿碟法)和漿式提取法[19]����。其中�����,USP<1724>收載3種型號的VDC和2種型號的浸沒池��,PMDA可以采用VDC和漿碟法進行試驗��,EMA則沒有明確說明�����。
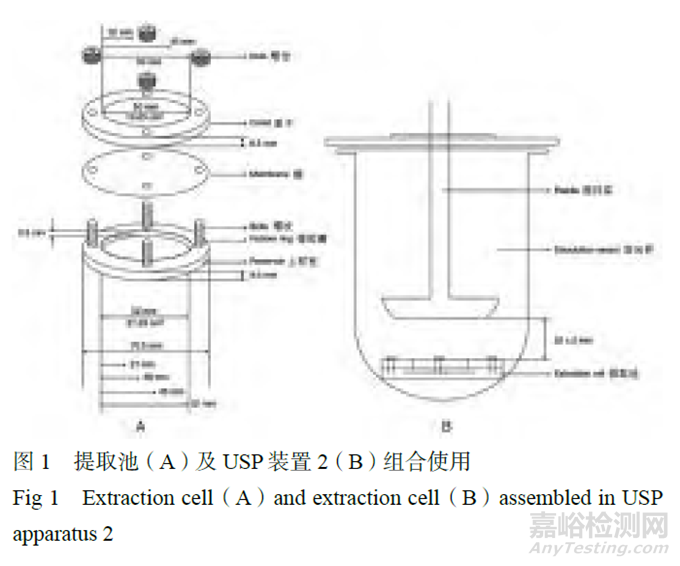
采用漿碟法時��,不需要使用膜����,因此需評估配方制劑溶解于接收介質的可能性[20]����。漿式提取法是將歐洲藥典(Ph. Eur)通則2.9.4中的提取池[21]與USP裝置2組合使用(見圖1)。雖然����,也可以采用浸沒池和流通池��,目前國內(nèi)外最常用的設備仍是VDC��。
對于VDC����,應根據(jù)實驗室的風險承受能力和生產(chǎn)商的建議�����,在6~12個月的周期內(nèi)對其進行再確認��,確認內(nèi)容包括但不限于:①確認供給室和接收室的孔口直徑在規(guī)定值的±5%����;②將所有組件組裝后測定接收室的體積(精確到0.01mL),在實際計算過程中�����,采用測定的體積替代標稱體積��;③溫度達到平衡后,確認膜表面溫度的穩(wěn)定性(在目標溫度的±1℃范圍內(nèi))�����;④攪拌速率(通常在400~600r·min-1)����。
在研究期間,應監(jiān)控和報告實驗室環(huán)境的溫度和濕度����。如適用����,建議將溫度控制在(21±2)℃、濕度控制在(50%±20%)�����,避免將設備置于空調(diào)的通風口之下及暴露在直射陽光中��。工作臺要保持水平��,建議傾斜度≤1°[22]��。
2.2 合成膜
除漿碟法和漿式提取法外,其他類型的擴散池都需要采用一種膜來防止接收介質的攪拌對樣品的機械擾動��。在IVRT研究中����,應選擇具有適當?shù)亩栊院蜕虡I(yè)化的人工膜(如聚丙烯膜、聚醚砜膜��、醋酸纖維素/尼龍混合酯膜�����、聚四氟乙烯膜�����、尼龍膜��、聚砜膜等)����,但不宜采用生物膜或模擬生物膜開發(fā)的合成膜,因為這些膜可能會不恰當?shù)赜绊懰幬镝尫诺谋碛^速率[4��,16]����。
EMA要求膜不應限制活性成分釋放��,PMDA要求膜呈非勻速滲透����。通常情況下�����,商業(yè)化的合成膜有不同的厚度��、孔徑和物理性質��,在IVRT研究中常用的膜厚度為70μm�����、孔徑為0.45μm�����。對于脂溶性藥物和親脂性基質����,可采用較大孔徑的膜(如1.2μm),此外可能還需要將膜預先在適當?shù)娜軇ㄈ缛舛罐⑺岙惐ィ┲薪?�,以賦予膜親脂性����,提高藥物從親脂性基質中擴散的速率。
纖維素膜在制造時通常會添加增塑劑(如甘油)�����,以保持膜的柔軟性��,使用前需去除增塑劑��,如可在接受介質中浸泡30min[23]����。在測試親脂性基質的軟膏時,使用預先浸泡的膜可以確保恒定的體外測試條件�����。此外����,接收介質和配方制劑不應影響膜的物理完整性��,Realdon等[24]使用不同類型的軟膏�����,在進行IVRT研究期間和之后采用“氣泡點(bubble point)”測試來評估膜的完整性����,這個測試也可以幫助決定是否需要對膜進行浸泡處理��。
膜的非勻速滲透��,可通過采用不同規(guī)格制劑(如50%�����、100%和150%規(guī)格)獲得不同的釋放速率進行評估�����。對于FDA和EMA要求的惰性(不與活性成分結合)評估方法為:①在IVRT的整個研究期間����,將膜放置到相應溫度的接收液中進行孵化(例如����,采用接收介質配制成待分析物的預期最高和最低濃度水平��,在(32±1)℃下放置6h)��,至少重復測定3份��;②并在相同條件下�����,同步考察3份不加膜的對照溶液�����,評估與膜吸附無關的藥物損失����;③如果實驗結束時����,接收液中藥物的回收率在(100±5)%,可認為在該條件下膜是惰性的����。
綜上所述�����,在合成膜的選擇過程中��,應選擇具有適當惰性和商業(yè)化的人工膜��,不宜采用生物膜或模擬生物膜開發(fā)的合成膜����。在方法開發(fā)過程中�����,應確定合成膜的種類(包括孔徑大小��、厚度��、材質)����、是否需要進行浸泡處理(如需浸泡處理,需確定浸泡介質和浸泡時間)����,同時需對膜的惰性和非勻速滲透情況進行評估。此外��,部分膜有正反面����,在試驗過程中,應根據(jù)廠家說明書進行使用����。
2.3 樣品分析方法
通常,IVRT樣品分析方法可基于成品的含量測定方法建立�����,但應確保所用的分析方法具有適當?shù)撵`敏度�����,以便可以定量測定從半固體制劑釋放到接收介質中低濃度水平的藥物量��。建議將接收液的選擇作為樣品分析方法開發(fā)的初始步驟����,其優(yōu)點是:在使用接收液對不同膜評估前,可先采用選定的接收液對樣品分析方法進行優(yōu)化����。此外應對方法的專屬性進行評估����,以確認從制劑和/或膜中浸出的其他物質不干擾藥物的定量測定�����;同時�����,應對樣品在最高相關溫度的接收液中放置的最長時間進行評估����。
根據(jù)FDA IVRT指南和USP<1724>,應采用多點校正曲線法(6~8個)進行測定�����。
2.4 IVRT方法開發(fā)
2.4.1 上樣量及上樣方式
FDA SUPAC-SS指南[16]建議上樣量為300mg����,在上樣過程中為防止樣品揮發(fā),供給室應保持封閉狀態(tài)。USP<1724>要求擬定的上樣量應確保劑量損耗不會影響試驗期間的穩(wěn)態(tài)藥物釋放動力學����,正如FDA IVRT指南所述�����,雖然一些外用制劑在劑量消耗大于30%時也能持續(xù)觀察到穩(wěn)態(tài)釋放動力學��,但通常而言�����,穩(wěn)態(tài)釋放動力學應假定劑量消耗低于30%��。EMA為偽無限上樣�����,但應確保實際上樣量在擬定上樣量的±5%范圍內(nèi)��。PMDA要求根據(jù)釋放和滲透的藥物量確定�����。
因此,通常建議上樣量在300mg以上��,以避免過量的劑量消耗��,或因上樣量過低導致上樣難度的增加(如需將上樣量控制在±5%范圍內(nèi))以及對檢測方法的重復性����、靈敏度造成挑戰(zhàn)。此外�����,EMA要求釋放量至少為標示量的70%����,PMDA要求至少為70%或達到平臺期,因此上樣量亦不能過高����,以免整個試驗時間過長。
除上述要求外����,在上樣過程中應盡量減少和控制剪切應力,采用相同的上樣方式(如直接從管中擠出到膜上�����、用抹刀轉移后涂覆到膜上、用外置活塞式移液器或一次性采集器轉移后分配)將藥物均勻地涂覆到膜上�����,制成緊湊的圓柱形�����,且在上樣過程中應避免膜與樣品之間產(chǎn)生氣泡�����。
綜上所述��,在IVRT方法開發(fā)過程中�����,應根據(jù)樣品特性的不同對不同的上樣方式進行評估,確保上樣的均勻性和一致性�����,并根據(jù)穩(wěn)定藥物釋放動力學、釋放量�����、試驗時長以及方法的靈敏度和重復性等要求綜合考慮確定樣品的上樣量��。
2.4.2 接收介質
Proniuk等[25]分別采用pH7.4磷酸鹽緩沖液(pH7.4PBS)和肉豆蔻酸異丙酯為接收介質����,用肉豆蔻酸異丙酯對膜進行浸泡�����,以及采用pH7.4PBS為接收介質并用pH7.4PBS對膜進行浸泡的三種方法����,對IVRT從兩種密切相關的水凝膠中選擇酮洛芬釋放速率最快的外用半固體制劑的能力進行評估����。結果表明,在產(chǎn)品開發(fā)早期進行處方篩選時�����,只有當接收介質具有與人體皮膚相似的特性時�����,IVRT才有助于預測可供吸收的藥物量�����,說明接收介質選擇的重要性����。
pH是接收介質選擇時需要考慮的一個重要因素�����,其選擇應基于配方制劑的pH�����、原料藥在不同pH條件下的溶解度以及藥物作用部位的pH,通常情況下�����,皮膚的pH在5~6[20,26]��。
雖然希望接收介質具有與皮膚相似的生理狀態(tài)��,但也必須確保其能夠恰當?shù)臏y量藥物的釋放����,而選擇的最重要因素是藥物在其中的溶解度��,即在半固體制劑的藥物釋放過程中����,接收介質應可以提供合適的漏槽條件����。
EMA指南[7]中未明確說明接收介質的組成,但要求在整個試驗期間����,接收介質中活性物質的最大濃度不應超過在該接收介質中活性物質最大溶解度的30%。同時要求����,應盡量減少接收介質的反向擴散,以避免藥物產(chǎn)品的轉化�����,且在整個釋放測試期間�����,應使接收介質的pH值保持恒定�����。
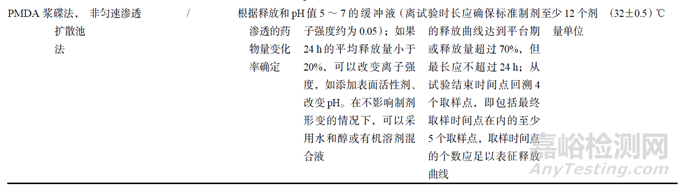
PMDA要求采用pH值在5~7的緩沖液(離子強度約為0.05)作為接收介質����,但當采用標準制劑進行試驗時��,如果24h的平均釋放量小于20%�����,則可改變離子強度��,如添加表面活性劑����、改變pH。在不影響制劑形變的情況下����,可以采用水醇或有機溶劑混合液。
FDA IVRT指南和USP<1724>指出接收介質通常為水和醇或有機溶劑混合液�����,要求藥物在接收介質中的溶解度應超過IVRT研究中最高樣品濃度����,理想情況下是一個數(shù)量級,同時需要證明在研究期間可達到穩(wěn)態(tài)釋放速率����。
根據(jù)上述分析�����,在接收介質選擇中�����,可從以下幾個方面進行考慮:①通常可采用水醇或有機溶劑混合液��,但采用該類型的接收介質時����,應盡量減少接收介質的反向擴散����,以避免藥物產(chǎn)品發(fā)生形變,如Kanfer等[27]研究表明乙醇可通過聚合物膜反向滲透導致乳膏制劑的完整性發(fā)生改變��;②對于水性緩沖液(pH值在5~7��,離子強度約為0.05)��,在整個釋放測試期間�����,應使接收介質的pH值保持恒定����,如釋放量較低�����,可添加表面活性劑�����、改變pH;③符合漏槽條件����;④采用標稱規(guī)格150%的配方制劑,也可達到穩(wěn)態(tài)釋放速率����;⑤在整個試驗期間,藥物在接收介質中穩(wěn)定��;⑥如適用��,需對接收介質進行脫氣處理����,避免試驗期間產(chǎn)生氣泡。
2.4.3 設備平衡
在IVRT試驗開始之前����,設備應在預定的試驗條件下進行適當?shù)钠胶狻MǔG闆r下需對接收介質進行脫氣處理����,以避免在試驗前或試驗過程中膜下產(chǎn)生氣泡�����。系統(tǒng)溫度應保持恒定并進行良好的控制����。根據(jù)不同的試驗方法����,IVRT膜可以在擴散池組裝之前預先用接收介質浸泡,也可以上樣前在擴散池內(nèi)與接收介質原位平衡(如在膜溫度為32℃下保持30min)�����。
2.4.4 試驗時長和取樣
根據(jù)USP<1724>��,試驗時長應適合于表征藥物的穩(wěn)態(tài)藥物釋放動力學�����,這通常是基于4~6h的持續(xù)穩(wěn)定釋放��。在第1h獲得的數(shù)據(jù)通常不能代表穩(wěn)態(tài)釋放動力學����,對于很多IVRT方法��,穩(wěn)態(tài)釋放動力學是根據(jù)1h之后的時間點計算的(例如�����,在1����、2��、3����、4和5h,或在2��、3�����、4����、5和6h)�����。至少選取5個取樣時間點�����,以獲得表征良好的釋放率��。在穩(wěn)態(tài)釋放速率評估中��,取樣時長至少為4h�����,較短的取樣時長(例如2h)可能不能有效表征穩(wěn)態(tài)釋放動力學。此外��,應精確控制樣品的取樣時間�����,將其控制在預定取樣時間的±15min或±2%��,取較窄的時間間隔范圍�����。FDA SUPAC-SS指南要求至少進行5次取樣�����。在取樣后����,需要采用預熱至系統(tǒng)預定溫度的接收介質進行補液�����,同時需確保�����,在每次取樣和補液后,膜下均無氣泡產(chǎn)生�����。
EMA要求試驗時長應足以表征釋放曲線�����,理想情況下����,釋放量至少為活性物質的70%。在藥物釋放曲線的線性部分至少選取6個時間點(每小時至少取樣一次)����,包括藥物擴散達到穩(wěn)態(tài)后的第一個取樣時間點。PMDA要求試驗時長應確保釋放曲線達到平臺期或釋放量超過70%��,但最長不應超過24h����。
如上所述,在試驗時長和取樣時間點方面��,各國要求存在差異,其中FDA僅要求對穩(wěn)態(tài)釋放動力學(即釋放曲線的線性部分)進行評估�����,EMA除需要對穩(wěn)態(tài)釋放動力學進行評估外�����,還要求在試驗結束時釋放量至少為70%�����,PMDA與EMA要求相似�����,但相對較為寬松(如釋放曲線達到平臺期或釋放量超過70%均可作為釋放終點)����。
因此�����,在IVRT研究過程中�����,對于試驗時長和取樣��,通常應滿足以下要求:①在穩(wěn)態(tài)藥物釋放階段至少選取5個取樣點(如申報歐盟����,至少選取6個時間點)����,且取樣時長不應少于4h;②將樣品的取樣時間控制在預定取樣時間的±15min或±2%����,取較窄的時間間隔范圍����;③如適用,釋放曲線應達到平臺期或釋放量超過70%��,但最長時間不應超過24h�����;④在取樣后,需采用預熱至系統(tǒng)預定溫度的接受介質進行補液��,同時需確保在每次取樣和補液后�����,膜下均無氣泡產(chǎn)生��。
2.4.5 重復次數(shù)
根據(jù)USP<1724>和FDA SUPAC-SS指南�����,通常采用6個劑量單位可足以表征該產(chǎn)品的性能(釋放速率),但在數(shù)據(jù)等效性評估中��,如不能滿足要求�����,應額外增加12個劑量單位進行評估����。EMA要求在首次進行方法驗證或等效性評估時,每個批次的樣品最少需要12個重復����,而在日常放行檢測中��,每個批次采用6個劑量單位即可��。PMDA主要用于上市后處方變更的生物等效性評估�����,要求每個批次至少采用12個劑量單位�����。
2.4.6 膜溫度
對于皮膚外用制劑��,F(xiàn)DA和EMA均要求控制在(32±1)℃�����,而PMDA要求將溫度控制在(32±0.5)℃����。對于直腸和陰道等內(nèi)用產(chǎn)品����,溫度應控制在(37±1)℃。如適用�����,實驗前用非接觸式紅外溫度計對溫度進行測定�����。
2.5 數(shù)據(jù)統(tǒng)計分析
對于每個擴散池����,應測定每個取樣點(t1�����,t2,etc)的藥物釋放量(通常以µg·cm-2為單位)��,并以累計釋放量對√t作圖����,在穩(wěn)態(tài)藥物釋放階段,兩者應呈線性關系(r2≥0.97),其斜率為藥物釋放速率��。在IVRT研究中��,各國均要求對藥物釋放速率的一致性進行評估�����。在理想情況下�����,EMA和PMDA還要求藥物的累計釋放量大于70%����。下文對FDA、EMA和PMDA指南中推薦的一致性評價方法進行介紹�����。
2.5.1 FDA
由于普遍存在測定誤差�����,如氣泡和膜缺陷�����,測定結果常呈非正態(tài)分布��,推薦采用Mann-Whitney U檢驗計算受試制劑(T)與參比制劑(S)之間斜率比值的90%置信區(qū)間��。通過以下示例說明��,采用6個劑量單位�����,分別以RS和TS代表參比制劑和受試批次的斜率�����,計算所有T/R斜率的比值�����,如表2所示�����。
在計算出T/R斜率比值后�����,將36個T/R斜率比值從低至高排序��。然后��,將第18個和第29個斜率比值轉換為百分單位��,分別代表90%置信區(qū)間的下限和上限����,如果兩者均在75%~133.33%內(nèi),則藥物釋放速率是等效的����。如果置信區(qū)間在75%~133.33%之外,R和T再各測12個劑量單位����,與第1次實驗結果組合,2組各有18個釋放速率�����,按上述進行置信區(qū)間計算�����,如果第110個和第215個斜率比值均在75%~133.33%內(nèi),可判定藥物釋放速率是等效的����。
2.5.2 EMA
與FDA的統(tǒng)計方法相似����,采用12個劑量單位,計算R和T各釋放速率的相互比值TS/RS(共計144個)��,然后對這些TS/RS值進行對數(shù)變換��,使用下式計算置信區(qū)間:
其中��,X是計算所得的所有TS/RS對數(shù)的平均值�����,n表示樣本量��,t是n-1個自由度(t)的90%置信區(qū)間的t值(n=6,t=2.015��;n=12��,t=1.796)��,s表示所有對數(shù)轉換比率的標準偏差�����。所得結果取反對數(shù)��,即為置信區(qū)間��。如果R和T釋放速率幾何均值比的90%置信區(qū)間在90%~111%內(nèi),則藥物釋放速率是等效的�����。
2.5.3 PMDA
在規(guī)定的試驗時間或釋放曲線達到平臺期后的1個時間點�����,以及和該時間點對應的釋放速率的一半所對應的時間點��,受試制劑與標準制劑的平均釋放速率之比應在0.8~1.2����。此外,PMDA要求受試制劑釋放速率的偏差與標準制劑相比�����,應相同(可采用F檢驗進行判斷)或更小�����。
3�����、結語
本文匯總了FDA��、EMA和PMDA等國家關于IVRT研究的政策法規(guī)�����,目前國內(nèi)陸續(xù)發(fā)布外用藥物制劑相關研究技術指南,但尚未有統(tǒng)一的技術規(guī)范對IVRT的研究進行規(guī)定�����。外用藥物制劑本身具有工藝復雜性,在IVRT研究中亦有很多技術難點�����,如合成膜的選擇及處理、接收介質的篩選����、上樣方式的控制等。伴隨著我國對外用制劑研究的越加深入����,希望結合上述對IVRT研究技術參數(shù)的討論����,重點參照2022年USP<1724>和FDA IVRT指南,結合我國實際需要�����,盡早統(tǒng)一IVRT研究技術要求����。