隨著歐盟MDR法規(guī)的實施推進�����,風(fēng)險管理流程與醫(yī)療器械的其他基本流程(如設(shè)計和開發(fā)�����、臨床評估��、上市后監(jiān)督等)之間的相互聯(lián)系正變得日益緊密��,而ISO/TR 24971正是使用風(fēng)險管理流程提高醫(yī)療器械質(zhì)量�����、安全性和有效性的寶貴工具���,被視為實施ISO 14971:2019的指南�����。
本期將深入探討ISO/TR 24971��,重點介紹有效的風(fēng)險管理流程實施的關(guān)鍵要求和方法��,該流程符合質(zhì)量管理體系法規(guī)(如ISO 13485�����、21 CFR 820)和相關(guān)的適用的法規(guī)要求(如歐盟MDR)��。
1���、ISO/TR 24971的風(fēng)險分析流程
■ 預(yù)期用途描述
■ 與安全有關(guān)的醫(yī)療器械特性
■ 識別與醫(yī)療器械相關(guān)的危害和危險
■ 評估風(fēng)險和危險情況
2、ISO/TR 24971中合理可預(yù)見的誤用
醫(yī)療器械的描述應(yīng)包含“合理可預(yù)見的誤用識別”的相關(guān)內(nèi)容�����。
誤用被定義為“以非制造商預(yù)期的方式使用醫(yī)療器械�����,但可能由易預(yù)測的人類行為導(dǎo)致”,其可能與使用錯誤�����、特定誤用或故意將醫(yī)療器械用于其他醫(yī)療目的有關(guān)�����,例如:超出預(yù)期用途���。
誤用可能由不同的原因引發(fā)�����,例如:對風(fēng)險認(rèn)知不佳�����、使用說明不夠清晰等��。
3、ISO/TR 24971與安全有關(guān)設(shè)備特征
為幫助制造商識別危險情況下的設(shè)備特征���,ISO/TR 24971 在附件A中提供完整的問題列表���,以支持識別可能影響安全的設(shè)備特征��。
4���、ISO/TR 24971識別危險的過程
識別危險是風(fēng)險分析過程的第一步。危險是潛在的傷害源�����,可能與醫(yī)療器械的使用或產(chǎn)品本身(設(shè)計)有關(guān)���。
通常��,醫(yī)療器械只有在發(fā)生一系列導(dǎo)致危險情況的事件時才會造成或?qū)е聜Α?/span>
與故障相關(guān)危險情況�����,可設(shè)想以下不同場景:
但當(dāng)未發(fā)生故障���,或可能與醫(yī)療器械相關(guān)特定療法具有內(nèi)在關(guān)聯(lián)時,也可能出現(xiàn)危險情況��。
5、ISO/TR 24971風(fēng)險評估的過程
1.每個已被識別的風(fēng)險都需要根據(jù)特定風(fēng)險的排名進行評估�����,評估內(nèi)容如下:
· 傷害發(fā)生的概率��;
· 傷害的嚴(yán)重程度��。
2.對發(fā)生概率的估計��,可使用2種不同的方法:定量和定性�����。當(dāng)具備充足的數(shù)據(jù)�����、具備充足的可信度���,用于估計傷害發(fā)生的概率時���,應(yīng)使用定量方法。否則��,定性方法的估計結(jié)果比具有高度不確定性的定量方法更為可信���。
應(yīng)高度關(guān)注無法估計發(fā)生概率的情況��,例如:軟件故障或誤用情況���。
當(dāng)無法估計傷害發(fā)生概率時,有必要僅根據(jù)傷害的嚴(yán)重程度作評估風(fēng)險��。
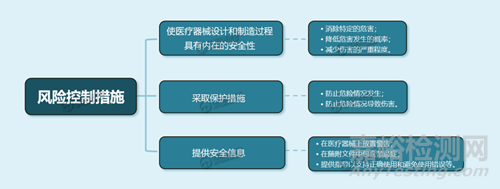
6��、ISO/TR 24971風(fēng)險控制的策略
實行風(fēng)險控制措施時�����,優(yōu)先順序至關(guān)重要���。
有關(guān)風(fēng)險控制選項的概述��,參閱以下方案:
7�����、ISO/TR 24971的總體剩余風(fēng)險評估
1.對整體剩余風(fēng)險的評估是風(fēng)險管理過程的重要時刻�����,包括總體剩余風(fēng)險評估���,即實行風(fēng)險控制措施后的風(fēng)險���。
2.ISO/TR 24971 提供不同的輸入,可用作評估剩余風(fēng)險的起點(包括但不限于以下情況):
· 不同的事件序列可能導(dǎo)致不同的危險情況和風(fēng)險���,每種都會導(dǎo)致整體剩余風(fēng)險��;
· 特定的傷害可能來自不同的危險情況���;
· 對醫(yī)療器械所有操作說明進行全面審查,可能發(fā)現(xiàn)說明的不一致或難以遵循�����。
3.ISO/TR 24971列出剩余風(fēng)險評估的不同方法示例:
· 與醫(yī)療器械預(yù)期用途相關(guān)的收益同整體剩余風(fēng)險進行權(quán)衡���;
· 剩余風(fēng)險的可視化表示���;
· 與市場上可用的類似醫(yī)療器械做比較�����;
· 聽取專家意見,以支持與醫(yī)療器械預(yù)期收益相關(guān)的總體剩余風(fēng)險評估�����。
注意:將所有單個剩余風(fēng)險相加的方式���,不可能用于總體剩余風(fēng)險評估���。評估總體剩余風(fēng)險并無首選方法,應(yīng)當(dāng)由制造商負(fù)責(zé)確定適當(dāng)?shù)姆椒ā?/span>