在問到一些新建企業(yè)的質(zhì)量管理體系建立進度時���,常常回答說:嗯,我們有**個文件�����,現(xiàn)在完成了**���,大約會在什么時候完成。��。�����。還有的會說�����,我們有某某企業(yè)的文件模板��,所以巴拉巴拉�����。以及:你有沒有某某文件的模板�����,能不能給我們巴拉巴拉��。
難道,質(zhì)量體系的建立��,只是完成GMP文件多少個嗎�����?
考核質(zhì)量體系的建立與否�����,只是有沒有完成文件體系嗎�����?
所以��,是不是應(yīng)該考慮一下質(zhì)量量度Quality metrics呢�����?一個成熟的質(zhì)量體系���,最終體現(xiàn)出來的���,當(dāng)然應(yīng)該是合格率的保證和持續(xù)提升�����、投訴退貨率的降低和持續(xù)下降,以及��,比如偏差和實驗室錯誤的發(fā)生率��、變更控制的效果與及時性���、產(chǎn)品回顧中的良好趨勢���、質(zhì)量風(fēng)險管理的有效、對產(chǎn)品生產(chǎn)和質(zhì)量控制的知識管理的不斷提升��。��。���。
那么�����,到底怎樣去看質(zhì)量體系的建立與否��、體系水平呢�����?
帶著這個問題���,最近�����,我又開始重新研究FDA的《藥品CGMP符合性的質(zhì)量體系方式》指南�����,重新理解質(zhì)量體系��,以及六大體系的關(guān)系:
1. 六大體系思路
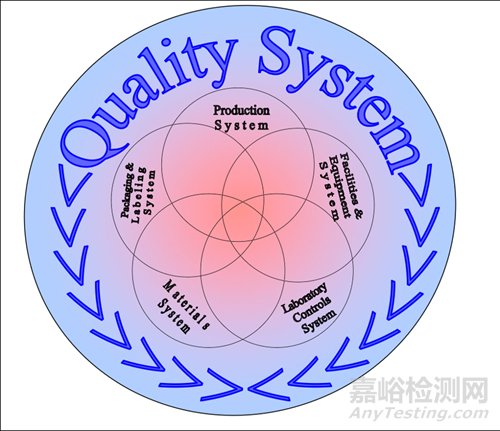
生產(chǎn)�����、設(shè)施設(shè)備��、實驗室控制��、物料�����、包裝貼簽五大體系涵蓋在質(zhì)量體系內(nèi)�����,它們之間也相互交聯(lián)��。這個意思�����,盯著這個圖中的幾個圈圈多看幾眼�����,自然就很容易理解���。
FDA提出的這六大體系思路,并不是建立質(zhì)量體系的指引���,而是FDA對藥品生產(chǎn)企業(yè)的檢查框架���。順著這個六大體系圖���,再去看FDA的CPGM7356.002,在每個體系的檢查重點前���,都有這樣一句話��,不厭其煩地重復(fù)了六次:
對以下內(nèi)容���,企業(yè)應(yīng)當(dāng)具有書面批準(zhǔn)的程序和由此產(chǎn)生的記錄。應(yīng)當(dāng)盡可能觀察核實企業(yè)是否遵守了這些書面程序�����。檢查領(lǐng)域不僅限于成品���,也可能包含組分和過程物料�����。這些領(lǐng)域可能表明不僅本系統(tǒng)存在缺陷�����,而且其它系統(tǒng)也存在需要擴大涵蓋范圍的缺陷��。檢查時除了質(zhì)量體系以外��,還選擇這個體系來檢查時���,下列內(nèi)容都應(yīng)當(dāng)涵蓋���,只不過涵蓋的深度可能因檢查結(jié)果而異。
FDA對工廠的檢查是基于風(fēng)險的方式��。他們把工廠管理分成六大體系��,除了質(zhì)量體系必檢以外��,其它體系根據(jù)具體的產(chǎn)品�����、風(fēng)險情況選擇性檢查��。所以���,他說如果選擇了檢查這個體系,那這下面的內(nèi)容都要檢查。如果發(fā)現(xiàn)問題���,不僅僅是這個體系有問題��,其它體系也可能存在相關(guān)的問題或者問題的原因���,可能需要擴大檢查范圍。
這�����,也是對圖形中那幾個圈圈之間涵蓋與互相交疊部分的實際說明���。五大體系的圈圈除了都在質(zhì)量體系內(nèi)以外���,任何一個圈圈都與另外四個圈圈全部有交疊。
2. 制劑生產(chǎn)企業(yè)的質(zhì)量體系
制劑企業(yè)的六大體系檢查程序��,在CPGM7356.002中(最近更新:2022年9月16日)�����。
在質(zhì)量體系的檢查內(nèi)容之前�����,有這樣一段話:
對質(zhì)量體系的檢查評估分兩個階段。第一階段是評估質(zhì)量部門是否有充分的職責(zé)去審核和批準(zhǔn)所有與生產(chǎn)��、質(zhì)量控制和質(zhì)量保證相關(guān)的程序并確保程序充分適合于其預(yù)期的用途�����,包括相關(guān)的記錄保存系統(tǒng)���;第二階段是評估收集的數(shù)據(jù)來發(fā)現(xiàn)質(zhì)量問題��。而這些質(zhì)量問題可能鏈接至檢查范圍內(nèi)的其它主要體系��。
制劑企業(yè)質(zhì)量體系的檢查重點���,包括16大方面:
1)對委外操作和物料供應(yīng)商的質(zhì)量監(jiān)管�����;
2)對質(zhì)量體系的建立���、實施��、監(jiān)督和持續(xù)改善管理(將質(zhì)量風(fēng)險管理與知識管理相結(jié)合)���;
3)質(zhì)量風(fēng)險管理程序��;
4)產(chǎn)品回顧��;
5)投訴審核�����;
6)差異與失敗調(diào)查��;
7)對所生產(chǎn)的所有產(chǎn)品的變更管理���;
8)批準(zhǔn)的申請產(chǎn)品的變更報告;
9)否決Rejects��;
10)穩(wěn)定性失?�?�;
11)隔離產(chǎn)品�����;
12)驗證;
13)cGMP人員的培訓(xùn)和資質(zhì)確認(rèn)��;
14)在整個產(chǎn)品生命周期內(nèi)持續(xù)監(jiān)測產(chǎn)品質(zhì)量和工藝性能的程序�����;
15)返工/再加工��;
16)退貨/補救
3. 原料藥生產(chǎn)企業(yè)的質(zhì)量體系
原料藥企業(yè)的六大體系檢查程序��,是在CPGM7356.002F中(最近更新時間2015年9月11日)��。
在質(zhì)量體系的檢查內(nèi)容之前�����,同樣也有類似的檢查的兩個階段的話:
同樣���,在質(zhì)量體系和其它每個體系的檢查重點之前���,也有要有書面程序�����、發(fā)現(xiàn)的缺陷可能關(guān)系到其它體系的缺陷等等這樣一段話:
原料藥企業(yè)質(zhì)量體系的檢查重點,包括14大方面:
1)確保實現(xiàn)質(zhì)量單位職責(zé)的充足的員工
2)定期的產(chǎn)品質(zhì)量回顧
3)投訴審核
4)與生產(chǎn)和檢測相關(guān)的差異和失敗調(diào)查
5)變更控制
6)退貨/補救
7)否決
8)原料放行系統(tǒng)
9)自上次檢查以來生產(chǎn)的批次���,以評估由于工藝問題而造成的否決或用途轉(zhuǎn)變(例如���,從藥用轉(zhuǎn)為非藥用)
10)返工/再加工
11)召回
12)穩(wěn)定性失敗
13)驗證
14)質(zhì)量控制單位職能的人員的培訓(xùn)和資質(zhì)確認(rèn)
對原料藥的QS檢查,參考規(guī)范是ICH Q7的以下章節(jié):
第2章:質(zhì)量管理
第13章:變更控制
第14章:物料否決和回用
第15章:投訴和召回
第16章:委托制造商(包括實驗室)
QS檢查重點�����,制劑企業(yè)的16條和原料藥企業(yè)的14條對比��,除了制劑的已批準(zhǔn)藥品的變更報告這個內(nèi)容更多的與美國的藥品監(jiān)管要求相關(guān)以外���,其它項目的不同���,有些是由于原料藥與制劑管理內(nèi)容或重要程度不同,有些�����,可能是因為程序更新時不夠完善�����。比如:
在制劑QS中列出但原料藥QS中未列出的:
制劑2)質(zhì)量體系的開發(fā)實施監(jiān)測完善,原料藥企業(yè)同樣需要重視��;
制劑3)質(zhì)量風(fēng)險管理程序���,原料藥企業(yè)也應(yīng)該重視��。風(fēng)險的直接影響不同�����,但風(fēng)險的識別管理相同��。原料藥的生命周期中實施質(zhì)量風(fēng)險管理�����,對于雜質(zhì)引入來說更是源頭管理��。
制劑11)隔離產(chǎn)品���,對原料藥管理來說應(yīng)該對應(yīng)的是防止差錯和混淆。
制劑14)在整個產(chǎn)品生命周期內(nèi)對工藝性能和產(chǎn)品質(zhì)量的持續(xù)監(jiān)測��,特別強調(diào)這一點�����,是強調(diào)要將出現(xiàn)的重大問題報告給高層管理者�����。而對原料藥來說�����,工藝性能和產(chǎn)品質(zhì)量的持續(xù)監(jiān)測�����,則通過產(chǎn)品回顧��、驗證���、變更控制來實現(xiàn)��,并持續(xù)改善���。
制劑1)對委外操作和物料供應(yīng)商的監(jiān)督管理,對原料藥來說也是需要管理的�����。而原料藥8)有個原料放行系統(tǒng),除了適當(dāng)性的評估后批準(zhǔn)放行以外�����,也可以關(guān)聯(lián)到供應(yīng)商的性能監(jiān)督活動中�����。
在原料藥QS中列出而制劑QS未列的:
原料藥1)確保實現(xiàn)質(zhì)量單位職責(zé)的充足員工���,對制劑來說也是重要的�����,也許這項內(nèi)容包含在了制劑2)質(zhì)量體系中��。
原料藥9)自上次檢查以來生產(chǎn)的批次中由于工藝問題造成的否決和轉(zhuǎn)為它用的情況���,對制劑來說,“否決”這項中已經(jīng)包括了�����,而轉(zhuǎn)為它用,對制劑來說基本不存在��,但是���,在實際工作中,是存在不合格的制劑轉(zhuǎn)給原料藥企業(yè)的可能性的���。