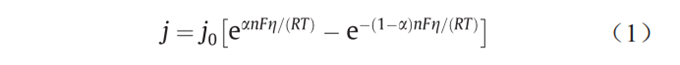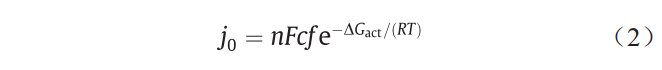在當(dāng)前能源需求和氣候變化的背景下���,可持續(xù)能源系統(tǒng)的研究已取得實(shí)質(zhì)性進(jìn)展���。可再生能源的大規(guī)模應(yīng)用需要高效的電能轉(zhuǎn)換和高密度的電能存儲(chǔ)技術(shù)���,以實(shí)現(xiàn)能源分配���。氫-水電化學(xué)轉(zhuǎn)化被視為一種理想的、無(wú)化石原料使用的可持續(xù)能源系統(tǒng)���,目前該能源系統(tǒng)中電解水和燃料電池兩種核心技術(shù)的能量轉(zhuǎn)換仍有較大改進(jìn)空間���,亟需進(jìn)一步實(shí)現(xiàn)技術(shù)突破���。
中國(guó)工程院院刊《Engineering》2020年6月刊發(fā)重慶大學(xué)魏子棟教授研究團(tuán)隊(duì)的《電化學(xué)氫-水轉(zhuǎn)化系統(tǒng)中電解水和氫燃料電池催化劑的設(shè)計(jì)》一文���。文章分析氫-水能源系統(tǒng)中電解水和燃料電池的能量耗散���,討論在催化劑表面發(fā)生的涉及氫-氧反應(yīng)的主要障礙,總結(jié)出催化活性趨勢(shì)的框架���,概述電化學(xué)氫-水轉(zhuǎn)化系統(tǒng)中的關(guān)鍵科學(xué)問(wèn)題���,提出開(kāi)發(fā)具有高能量轉(zhuǎn)化率的催化劑的研究方向���,為高活性氫-氧反應(yīng)電催化劑的設(shè)計(jì)提供思路。
一���、引言
目前全球能源消耗持續(xù)增長(zhǎng),但近88%的能源經(jīng)濟(jì)依賴化石燃料���。盡管化石燃料在能源組合中所占份額過(guò)大���,但化石燃料時(shí)代或許即將結(jié)束?��;剂洗罅肯幕驘o(wú)利可圖的開(kāi)采只是時(shí)間問(wèn)題���。除了供應(yīng)量減少的問(wèn)題外,化石燃料的使用對(duì)全球生態(tài)系統(tǒng)構(gòu)成了重大風(fēng)險(xiǎn)���。目前,世界上的能源供給主要依靠化石燃料的燃燒。這種燃燒所產(chǎn)生的副產(chǎn)品嚴(yán)重污染空氣���、土壤和水���。我們迫切需要采用新的思維方式,以便找到解決這些問(wèn)題的方案���,并設(shè)計(jì)更安全���、可持續(xù)的能源供應(yīng)系統(tǒng)���?��?稍偕茉磳⒃谑澜缒茉次磥?lái)中發(fā)揮至關(guān)重要的作用���。然而���,可再生能源和傳統(tǒng)能源之間的巨大差異造成了市場(chǎng)壁壘���?��?稍偕茉串a(chǎn)生的能量可能在短時(shí)間內(nèi)發(fā)生不可預(yù)測(cè)的變化���。例如���,太陽(yáng)能系統(tǒng)僅在陽(yáng)光照射時(shí)產(chǎn)生能量���。其他可再生能源,如風(fēng)能和潮汐能���,同樣具有不穩(wěn)定性這一不利因素���。這種不穩(wěn)定性使當(dāng)前的可再生能源發(fā)電的可靠性低于化石燃料產(chǎn)生的能源���,因?yàn)槠漭敵龈叨纫蕾囉谔鞖鈼l件和時(shí)間。為了使可再生能源大規(guī)模應(yīng)用���,需要高效的電能轉(zhuǎn)換和高密度的電能存儲(chǔ)技術(shù),以實(shí)現(xiàn)能源分配���。
電化學(xué)氫-水轉(zhuǎn)化(H2+O2 ? H2O)是一種清潔高效的可持續(xù)能源系統(tǒng)執(zhí)行解決方案。具體來(lái)說(shuō)���,可再生能源可以通過(guò)水電解轉(zhuǎn)化為儲(chǔ)存在氫氣中的化學(xué)能���。相反���,氫分子可以通過(guò)電化學(xué)的方式重組成水���,以便通過(guò)燃料電池輸出電能���。在該能量系統(tǒng)中���,氫充當(dāng)能量載體���,并且能量轉(zhuǎn)換與熱循環(huán)無(wú)關(guān)���。因?yàn)樵撓到y(tǒng)是基于電化學(xué)反應(yīng)���,可以有效地避免對(duì)自然環(huán)境和人類健康有害的氣體和化合物的釋放。
然而���,在實(shí)際應(yīng)用中���,氫氣必須首先被獲取���,然后被儲(chǔ)存,最后被轉(zhuǎn)換回水以釋放儲(chǔ)存的能量���。為了實(shí)現(xiàn)這一目標(biāo)���,高效、低成本的水電解和燃料電池技術(shù)必須有效地結(jié)合起來(lái)���。電化學(xué)過(guò)程是這些能量轉(zhuǎn)換技術(shù)的核心���,包括水電解技術(shù)中的析氫反應(yīng)(HER)和析氧反應(yīng)(OER)���,氫氧燃料電池中的氧還原反應(yīng)(ORR)和氫氧化反應(yīng)(HOR)���。這四種電化學(xué)反應(yīng)的效率對(duì)上述能量轉(zhuǎn)換技術(shù)的輸出性能有很大的影響。因此���,在該可持續(xù)能源系統(tǒng)中���,最關(guān)鍵的問(wèn)題是如何在催化電極表面有效地催化以上反應(yīng)���,以獲得最低的過(guò)電位和最高的電流密度���。除了電化學(xué)反應(yīng)引起的電壓降外,內(nèi)阻和傳質(zhì)電阻等也會(huì)影響水電解和燃料電池的總電壓���。因此���,通過(guò)優(yōu)化電極結(jié)構(gòu)來(lái)加速電子���、質(zhì)子的轉(zhuǎn)移和產(chǎn)物的脫附是另一個(gè)需要關(guān)注的問(wèn)題���。
文章對(duì)用于電化學(xué)氫-水轉(zhuǎn)化電催化劑的結(jié)構(gòu)工程的最新進(jìn)展進(jìn)行了全面綜述���。主要討論了兩個(gè)問(wèn)題:① 電化學(xué)氫-水轉(zhuǎn)換系統(tǒng)能量耗散的來(lái)源���;② 基礎(chǔ)科學(xué)與實(shí)用技術(shù)相結(jié)合驅(qū)動(dòng)的高能量轉(zhuǎn)換效率電催化劑的結(jié)構(gòu)設(shè)計(jì)���。在簡(jiǎn)要介紹了氫-水轉(zhuǎn)化過(guò)程中的電化學(xué)過(guò)程之后���,我們從實(shí)用的角度回顧了水電解和燃料電池兩種功能技術(shù)的能量耗散���,并利用經(jīng)典動(dòng)力學(xué)分析了催化劑表面發(fā)生電化學(xué)反應(yīng)的關(guān)鍵障礙���。借助于反應(yīng)中間體之間的標(biāo)度關(guān)系���,我們構(gòu)建了一個(gè)了解催化性能趨勢(shì)的框架���,為開(kāi)發(fā)用于廣泛反應(yīng)的高效催化劑提供指導(dǎo)���?��?偨Y(jié)了設(shè)計(jì)高性能電催化劑的通用策略,并討論了它們的優(yōu)缺點(diǎn)���。這部分介紹了通過(guò)結(jié)構(gòu)設(shè)計(jì)實(shí)現(xiàn)的高效電催化劑的典型案例���,展示了合成化學(xué)、電催化化學(xué)和計(jì)算化學(xué)的有機(jī)結(jié)合���。最后概述了電化學(xué)氫水轉(zhuǎn)化系統(tǒng)中的關(guān)鍵科學(xué)問(wèn)題���,為高效、可再生能源系統(tǒng)的催化劑設(shè)計(jì)提供了方向���。
二、電催化基礎(chǔ)
(一) 氫-水轉(zhuǎn)化中的電化學(xué)反應(yīng)
如圖1所示���,該可再生清潔能源系統(tǒng)涉及兩種不同的功能技術(shù):電解水和燃料電池���。這兩種技術(shù)的電解槽主要由四部分組成:電解液(如H2O)���、離子交換膜(如Nafion膜)、陽(yáng)極電極和陰極電極���。為了提升這兩種技術(shù)的效率,陰陽(yáng)電極表面一般涂覆著高效穩(wěn)定的催化劑層���。在水電解槽中���,電能被消耗以將水分解為氣態(tài)氫(H2)和氧(O2)。以酸性水電解為例���,當(dāng)電子通過(guò)外回路時(shí)���,質(zhì)子通過(guò)離子交換膜進(jìn)入陰極,與電子結(jié)合形成氫分子���,水在陽(yáng)極氧化形成氧分子和質(zhì)子。
圖1 氫-水的電化學(xué)轉(zhuǎn)換中電解水和燃料電池的反應(yīng)原理圖
燃料電池中發(fā)生的電化學(xué)反應(yīng)與水電解過(guò)程完全相反���。氫和氧的自然“冷”燃燒發(fā)生在燃料電池裝置中���,其中氫作為燃料,氧作為氧化劑���。通常���,氫通過(guò)電極孔擴(kuò)散到陽(yáng)極表面。通過(guò)催化劑層的催化作用���,吸附的氫被電離并在電極上釋放出一個(gè)電子���。接下來(lái),通過(guò)電解液的氫離子和通過(guò)外部電路的電子都到達(dá)陰極���,與氧分子重新結(jié)合形成水分子���,并釋放出電流。通過(guò)使用合適的水或空氣冷卻系統(tǒng)���,可以消除內(nèi)部反應(yīng)和電阻產(chǎn)生的熱量���。表1總結(jié)了水電解和燃料電池在不同介質(zhì)中的半電池反應(yīng)。這四個(gè)反應(yīng)可分為兩個(gè)可逆反應(yīng)對(duì):相對(duì)于可逆氫電極(RHE)���,平衡電位(U0)為0 V的HER和HOR���,以及U0 =1.23 V的ORR和OER。從化學(xué)角度來(lái)看���,氫-水轉(zhuǎn)化是由兩個(gè)氧化還原對(duì)組成的:高電位的水/氧對(duì)和相對(duì)低電位的水/氫對(duì)。在酸性介質(zhì)中���,水合質(zhì)子將電荷從陽(yáng)極轉(zhuǎn)移到陰極,而在堿性電解質(zhì)中���,氫氧化物離子作為電荷載體從陰極轉(zhuǎn)移到陽(yáng)極���。
表1 在酸性和堿性電解質(zhì)中電解水和燃料電池的半電池反應(yīng)
(二)水電解和燃料電池的能量耗散
根據(jù)氫-水轉(zhuǎn)化的反應(yīng)熱力學(xué)���,水電解和氫氧燃料電池在標(biāo)準(zhǔn)條件下具有相同的起始電位(1.23 V)。然而���,這兩種技術(shù)的實(shí)際起始電位與標(biāo)準(zhǔn)電位相差甚遠(yuǎn)。在實(shí)際操作條件下���,即使使用最先進(jìn)的貴金屬作為電催化劑���,燃料電池的電壓始終低于0.9 V���,而水電解高于1.8 V���。在實(shí)際應(yīng)用中���,為了驅(qū)動(dòng)電化學(xué)反應(yīng)過(guò)程���,必須克服許多勢(shì)壘,包括電路的電阻���、電化學(xué)反應(yīng)的活化能���、產(chǎn)物氣泡或水對(duì)電極表面的堵塞,以及電解質(zhì)溶液的離子轉(zhuǎn)移電阻等���。這些勢(shì)壘需要足夠的電能供應(yīng)以克服,這大大降低了能量轉(zhuǎn)換效率���,導(dǎo)致工作電位低于熱力學(xué)電位���,即發(fā)生所謂的極化現(xiàn)象���。
圖2示出了典型的液體電解質(zhì)電池中的電阻(即勢(shì)壘)���。兩端的第一個(gè)電阻是外部電路電阻,包括陽(yáng)極和陰極的導(dǎo)線和連接的電阻以及電子穿過(guò)催化劑層的電阻���。在水電解中���,由于產(chǎn)生的氣泡對(duì)電極表面的覆蓋���,阻礙了電極與電解液的接觸���。類似地,產(chǎn)物水在電極與反應(yīng)物氧氣之間起到阻塞作用���,導(dǎo)致堿性燃料電池(AFC)中的傳質(zhì)阻力���。
圖2 (a)典型的液態(tài)堿性燃料電池示意圖;(b)電解池的橫截面���;(c)等效電路
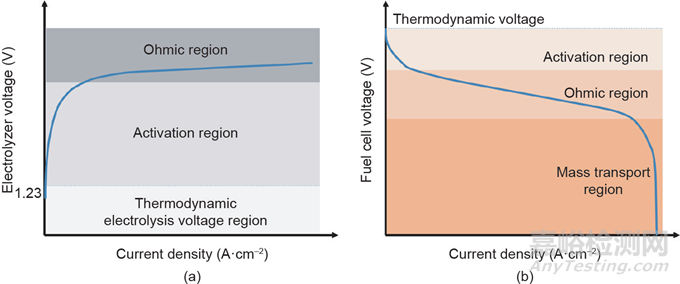
以上電池系統(tǒng)中的電阻可分為三類:活化電阻(電化學(xué)反應(yīng)引起的損耗)、歐姆電阻(離子和電子傳導(dǎo)引起的損耗)和濃度電阻(物質(zhì)傳輸引起的損耗)���。這三類電阻共同決定了電化學(xué)電池的電流-電壓(i-V)曲線的特征形狀���。圖3顯示了水電解和燃料電池的典型i-V曲線���。在水電解的i-V曲線中[圖3(a)]���,當(dāng)電壓高于熱力學(xué)電解電壓1.23 V后,電流開(kāi)始流過(guò)電解池���。在低電流密度下,歐姆電阻引起的電壓降很小���,反應(yīng)活化過(guò)電壓占電壓降的主要部分。此時(shí)極化曲線的對(duì)數(shù)形狀(Tafel區(qū)域)歸因于陽(yáng)極和陰極的電荷轉(zhuǎn)移現(xiàn)象���。隨著過(guò)電壓的進(jìn)一步增大,反應(yīng)活化勢(shì)壘減小���,極化曲線呈線性���。這種線性形狀表明此時(shí)歐姆電阻是電解池的最關(guān)鍵動(dòng)力學(xué)參數(shù)。在燃料電池的i-V曲線中[圖3(b)]���,活化電阻主要影響曲線的初始部分���,歐姆電阻的影響主要在曲線的中間部分���,濃度電阻的影響在曲線的尾部顯著���。盡管這兩種功能技術(shù)中發(fā)生的反應(yīng)是可逆的,但i-V曲線的形狀并不相同:水電解的i-V曲線在高電位下通常遵循Butler-Volmer模型���;而由于對(duì)傳質(zhì)速率的限制���,燃料電池的i-V曲線往往在高電位下顯示恒定值���。
圖3 水電解(a)和燃料電池(b)的極化曲線圖
為了提高兩種技術(shù)的能量轉(zhuǎn)換效率以改善能源系統(tǒng)的整體性能,必須了解上述電阻產(chǎn)生的緣由���,以便將其最小化���。歐姆損耗是由電極材料對(duì)電子流的電阻和電解液對(duì)離子流的電阻引起的���。這些損耗可以通過(guò)使用高導(dǎo)電材料作為布線和電極基板,以及通過(guò)減小兩個(gè)電極之間的距離來(lái)降低���。通過(guò)增加氣態(tài)反應(yīng)物的壓力或液體電解質(zhì)的濃度,可以減輕由傳質(zhì)引起的損耗���。這兩種電壓降主要取決于電池的設(shè)計(jì)和運(yùn)行條件���。除了上述兩類電阻外���,電化學(xué)電池中的大部分電壓降(>60%)是由半電池反應(yīng)的吉布斯自由能變化引起的。根據(jù)反應(yīng)的方向���,活化極化大大增加發(fā)生氧化反應(yīng)的陽(yáng)極電壓���,并降低了發(fā)生還原反應(yīng)的陰極電壓。
在電化學(xué)中���,Butler-Volmer關(guān)系被用作主要出發(fā)點(diǎn)���,將催化劑-電解質(zhì)界面上的過(guò)電壓(η)與該界面上的電流密度j(A·cm–2)聯(lián)系起來(lái):
式中,η是過(guò)電壓���,即界面上的實(shí)際電壓與平衡電壓之差���;j0是交流電流密度���,單位為A·cm–2���;α是電荷轉(zhuǎn)移的系數(shù)���;n是在電化學(xué)反應(yīng)中轉(zhuǎn)移的電子數(shù)���;F≈96485 C·mol–1,是法拉第常量;R是摩爾氣體常量(0.082 J·K–1·mol–1)���;T是熱力學(xué)溫度(K)���。Butler-Volmer方程表明���,電化學(xué)反應(yīng)產(chǎn)生的電流隨過(guò)電壓和交換電流密度呈指數(shù)增加���。實(shí)際上���,j0表示反應(yīng)物與產(chǎn)物處于平衡狀態(tài)時(shí)的“交換速率”���,是提高反應(yīng)速率的重點(diǎn)���。為簡(jiǎn)單起見(jiàn)并考慮濃度效應(yīng)���,正向反應(yīng)下j0的定義為:
式中,c是反應(yīng)物的表面濃度;f是產(chǎn)物的衰減率���;ΔGact是正向反應(yīng)的活化勢(shì)壘。等式(2)清楚地表明���,在給定的環(huán)境條件下���,減小活化能壘(ΔGact)將增加j0���。在實(shí)際反應(yīng)中���,只有處于活化狀態(tài)的物質(zhì)才能經(jīng)歷從反應(yīng)物到產(chǎn)物的轉(zhuǎn)變���。催化電極是物質(zhì)活化和轉(zhuǎn)變的場(chǎng)所,反應(yīng)的活化能極大程度上取決于電極材料���。因此使用高性能的催化電極可顯著降低反應(yīng)的活化勢(shì)壘���,進(jìn)而顯著提高j0���。基于對(duì)活化能���、電極材料和表面構(gòu)型之間關(guān)系的理解���,現(xiàn)今大量的研究工作集中于高效催化電極材料的設(shè)計(jì)以降低電極反應(yīng)的活化能。
(三)用于電化學(xué)氫-水轉(zhuǎn)化的催化劑設(shè)計(jì)指南
根據(jù)反應(yīng)機(jī)制���,反應(yīng)活化勢(shì)壘(ΔGact)可以用平衡電勢(shì)下速率決定步驟(RDS)的自由能變化(ΔGmax)來(lái)定量���,并且其在不同催化材料上的理論值可以通過(guò)密度泛函理論(DFT)來(lái)計(jì)算���。通過(guò)繪制j0與ΔGmax之間的火山曲線���,可以建立活化能與電極材料之間的關(guān)系���。最常見(jiàn)的火山曲線是基于Langmuir吸附類型的析氫反應(yīng)速率描述,其最大值位于氫吸附自由能(ΔGH*)為零附近���。在析氫反應(yīng)中���,反應(yīng)物首先吸附在催化劑表面形成反應(yīng)中間體(M-Hads)。在上述Volmer步驟之后���,氫分子可以由電解液中電子和質(zhì)子通過(guò)Heyrovsky步驟耦合形成���,或者通過(guò)Tafel步驟直接結(jié)合形成���。因此,氫吸附自由能(ΔGH*)是析氫反應(yīng)速率的決定性因素���。
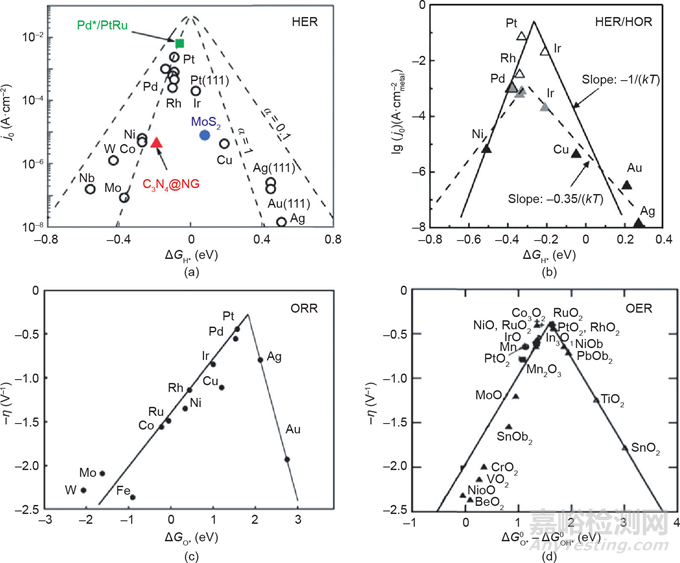
近年來(lái)���,DFT計(jì)算得到的材料表面的氫吸附自由能被廣泛用作許多傳統(tǒng)金屬和非金屬催化材料的活性描述符。如圖4(a)所示���,不同金屬的析氫交換電流密度存在顯著差異���,位于火山曲線頂部附近的金屬(如Pt)具有最佳的ΔGH*。如果催化材料對(duì)氫的吸附力較弱���,氫原子在材料表面幾乎不能被吸附���,整個(gè)反應(yīng)速率由氫的吸附步驟(Volmer步驟)決定。反之,氫原子在催化材料上的吸附太強(qiáng)���,M–Hads鍵則很難被打斷而形成H2���,反應(yīng)決速步為解吸步驟(Heyrovsky/Tafel)。作為HER的逆過(guò)程���,HOR的RDS是H2在催化劑表面的解離吸附���,它涉及電子從催化劑表面轉(zhuǎn)移到H2分子的反鍵軌道(σ*軌道)因此,M-Hads的強(qiáng)弱在HOR的動(dòng)力學(xué)中也起主導(dǎo)作用���,并且由于這兩個(gè)反應(yīng)的高度可逆性,HOR在貴金屬表面上的活性趨勢(shì)與HER相同[圖4(b)]���。
圖4 (a)各種材料表面HER交換電流密度(j0)和ΔGH*的關(guān)系圖���;(b)酸性介質(zhì)中表面歸一化的HOR/HER交換電流密度(j0)對(duì)ΔGH*的火山曲線圖;(c)不同金屬的ORR活性和ΔGO*的關(guān)系圖���;(d)氧化物的OER活性對(duì)ΔGO*–ΔGOH*圖
與氫參與反應(yīng)相似���,j0和ΔGmax之間的關(guān)系也應(yīng)用于氫-水轉(zhuǎn)化過(guò)程中氧參與的反應(yīng)���。如圖4(c)、(d)所示���,除了決定反應(yīng)速率的反應(yīng)中間體不同���,氧參與反應(yīng)的火山曲線形狀非常相似。ORR包括以四電子途徑將氧還原為水���,或以兩電子途徑生產(chǎn)過(guò)氧化氫���。事實(shí)上,一個(gè)直接的四電子ORR反應(yīng)機(jī)制可以是一個(gè)解離或結(jié)合過(guò)程���,這取決于催化劑表面的氧解離能壘���。因此,ORR火山曲線以氧吸附自由能(ΔGO*)與催化活性相關(guān)聯(lián)[圖4(c)]���。對(duì)于與氧結(jié)合太強(qiáng)的金屬���,ORR反應(yīng)速率受到O*或OH*物種脫附的限制���。對(duì)于與氧結(jié)合太弱的金屬,反應(yīng)速度可能受O2中O–O鍵分裂的限制(解離機(jī)制)���,或者受電子和質(zhì)子轉(zhuǎn)移到吸附的O2的限制(結(jié)合機(jī)制)���,具體情況取決于外加電位。如圖4(c)所示���,即使鉑(Pt)也不在絕對(duì)峰值���,ΔGO*略低于鉑的金屬應(yīng)具有更高的氧還原活性。根據(jù)上述熱力學(xué)火山曲線���,Nørskov等考慮到氫氧結(jié)合能是變化的,建立了ORR的微觀動(dòng)力學(xué)模型���。他們發(fā)現(xiàn)了一種與熱力學(xué)活性火山曲線非常一致的動(dòng)力學(xué)火山曲線���,并確定了ΔGO*比Pt(111)弱0.1 eV的催化劑具有最佳的四電子ORR活性���。
OER火山曲線始于1984年,具有很長(zhǎng)的歷史���。當(dāng)時(shí)Trasatti用金屬氧化物中金屬?gòu)牡脱趸瘧B(tài)到較高氧化態(tài)的轉(zhuǎn)變焓來(lái)描述氧化物電極的OER電催化活性���。這項(xiàng)開(kāi)創(chuàng)性的工作將OER過(guò)程視為表面配位化合物的兩種不同構(gòu)型之間的過(guò)渡。因此���,所有難氧化或易氧化的金屬氧化物對(duì)OER催化都不是很活潑���。難氧化意味著中間體的吸附性較弱,水離解是RDS���。反之���,易氧化性表明中間產(chǎn)物具有很強(qiáng)的吸附性,則O*或OH*的脫附為RDS���。因此���,與ORR相同���,OER反應(yīng)速率與ΔGO*有關(guān)。
然而���,四電子OER涉及多個(gè)中間體(OOH*���、OH*和O*),它們的結(jié)合能緊密相關(guān)且?guī)缀醪唤怦?��,?Delta;GO*作為OER活性的單一描述符并不準(zhǔn)確���。不同的反應(yīng)中間體的結(jié)合能之間存在線性標(biāo)度關(guān)系,即如果與一個(gè)反應(yīng)步驟相關(guān)的能量發(fā)生變化���,其他反應(yīng)步驟的能量也發(fā)生變化���。Man等將兩個(gè)中間產(chǎn)物(ΔGO* – ΔGOH*)的結(jié)合能之差作為化合物(包括金紅石、鈣鈦礦���、尖晶石、巖鹽和方鐵錳礦氧化物)催化活性的描述符[圖4(d)]���,其活性很好地服從火山曲線���。在金屬氧化物材料中���,各吸附位點(diǎn)的OH*和OOH*結(jié)合能(無(wú)論是在OER還是ORR中)均以大約3.2 eV的恒定能量值相互關(guān)聯(lián)。由于OOH*和OH*之間的非理想標(biāo)度���,即使對(duì)于OER和ORR火山曲線頂部的催化材料���,催化劑也存在最小理論超電勢(shì)(0.3~0.4 V),包括最優(yōu)的RuO2析氧催化劑和Pt基ORR催化劑���。
火山曲線恰當(dāng)?shù)刈C明了Sabatier理論���,即理想的催化劑與反應(yīng)中間體結(jié)合既不太弱也不太強(qiáng)。換言之���,對(duì)反應(yīng)中間體具有適當(dāng)結(jié)合能的催化劑表面可實(shí)現(xiàn)最佳催化活性���。具體來(lái)說(shuō),最理想HER/HOR催化劑是具有最小ΔGH*絕對(duì)值的材料���,理想ORR和OER催化劑則具有優(yōu)異的ΔGO*和ΔGO* – ΔGOH*���。除了降低活化勢(shì)壘外���,還有另一個(gè)顯著的方法來(lái)增加j0,即增加單位面積內(nèi)反應(yīng)位點(diǎn)的數(shù)量���。j0表示單位面積的反應(yīng)電流���,而電流密度的面積通常基于電極的幾何面積���。表面極其粗糙的電極的真實(shí)電極表面積可以比其幾何電極面積大幾個(gè)數(shù)量級(jí)���,因此可以提供更多的反應(yīng)位點(diǎn)。因此���,粗糙電極表面的有效j0將遠(yuǎn)大于光滑電極表面的有效j0���。提高活性位點(diǎn)密度的另一個(gè)簡(jiǎn)單方法是增大給定電極上催化劑的用量���。然而���,過(guò)量的催化劑會(huì)阻礙電極表面的電荷和質(zhì)子轉(zhuǎn)移���。因此���,電極的活性不隨催化劑用量的增加而線性增加。
總之���,提高電催化劑體系的活性(或反應(yīng)速率)一般有兩種策略:① 提高每個(gè)活性位點(diǎn)的本征活性���;② 提高給定電極上活性位點(diǎn)的數(shù)量。兩種方法各有利弊���。不同催化劑的本征活性差異可能超過(guò)10個(gè)數(shù)量級(jí)���,而由催化劑負(fù)載引起的活性差異僅為1~3個(gè)數(shù)量級(jí)。提高每個(gè)活性位點(diǎn)的本征活性是實(shí)現(xiàn)高活性的最根本���、最有效的途徑���,但其實(shí)現(xiàn)必須建立在對(duì)反應(yīng)機(jī)制和材料性能深入了解的基礎(chǔ)上���。增加活性位點(diǎn)的數(shù)量是一個(gè)更簡(jiǎn)單的策略,但活性增長(zhǎng)是有限的���。同時(shí)���,通過(guò)提高催化劑負(fù)載量來(lái)提高活性需要以增加電極成本和傳質(zhì)阻力為代價(jià)。在實(shí)際應(yīng)用中���,以上兩種策略可以同時(shí)實(shí)施���,從而大大提高催化劑的活性。
三���、電化學(xué)氫–水轉(zhuǎn)化中催化材料設(shè)計(jì)
(一)納米構(gòu)筑
眾所周知���,催化劑的電流密度隨著活性中心密度的增加而增加。暴露更多的活性中心對(duì)獲得高催化活性十分重要���。納米構(gòu)筑被認(rèn)為是提高活性位點(diǎn)密度最直接有效的策略���。在過(guò)渡金屬合金體系中���,人們首先認(rèn)識(shí)到實(shí)際活性表面積與電催化劑整體性能之間的關(guān)系。
早在1982年���,Brown等發(fā)現(xiàn)合金表面通常比單一金屬表面粗糙,可以為催化反應(yīng)提供更多的活性中心���。借助于Ni-Mo合金納米結(jié)構(gòu)化和鉬的選擇性腐蝕���,Ni-Mo合金的表面積大大增加,催化活性明顯提高���。
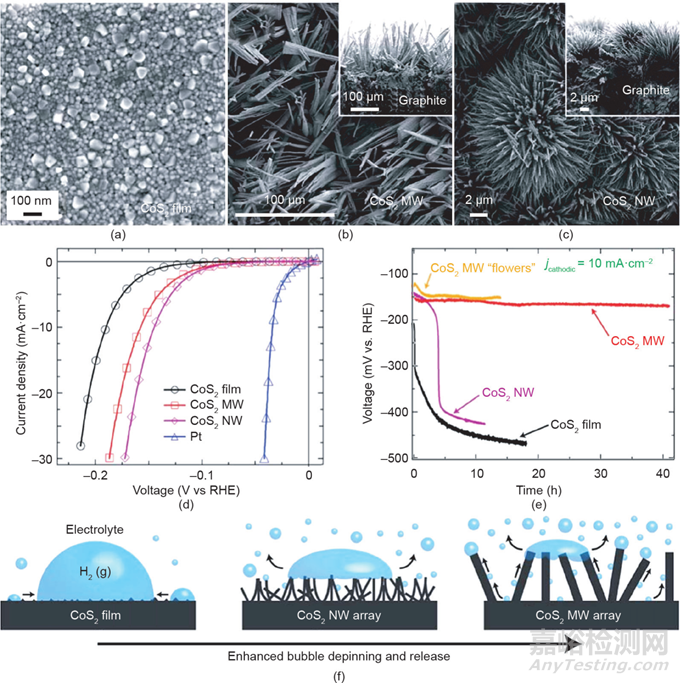
近十年來(lái)���,隨著合成技術(shù)的快速發(fā)展,一系列不同形貌的電催化納米材料相繼問(wèn)世���,包括納米籠���、納米纖維、納米花、納米泡沫���、納米網(wǎng)���、納米針、納米環(huán)���、納米殼���、納米線等。Faber等報(bào)道了金屬二硫化鈷(CoS2)作為一種高活性催化劑���,并證明了幾何結(jié)構(gòu)在決定其整體催化性能中的關(guān)鍵作用���。與常見(jiàn)的納米顆粒和納米薄膜形貌的電極相比,微納米結(jié)構(gòu)電極的高活性比表面積顯著改善了其催化性能(圖5)���。因此CoS2納米線電極只需低至145 mV的過(guò)電位以驅(qū)動(dòng)–10 mA·cm–2的析氫電流密度���。此外,通過(guò)促進(jìn)物質(zhì)傳遞和產(chǎn)物(氣泡或水)從催化劑表面的脫除���,納米結(jié)構(gòu)具有提高操作穩(wěn)定性和反應(yīng)速率的雙重功能���。Peng等使用自組裝和預(yù)成形策略���,可控合成了具有二維(2D)層狀結(jié)構(gòu)的Mo2C/C催化材料。高分散的Mo2C納米顆粒及二維層狀結(jié)構(gòu)有效地促進(jìn)了Mo2C活性中心的質(zhì)子和電荷轉(zhuǎn)移���,促進(jìn)了電化學(xué)HER過(guò)程。此外���,我們還進(jìn)一步合成了一系列三維納米催化材料���,包括NiCo2(SOH)x納米花、珊瑚狀FeNi(OH)x���、Ni-VC納米叢���、Ni-Mo2C納米線和Ni(OH)2@Ni2P納米柱。所有這些材料都具有高活性表面���、快速電子轉(zhuǎn)移和氣體逸出通道���,有利于催化水電解反應(yīng)進(jìn)行���。
圖5 (a)~(c)不同形貌CoS2的掃描電鏡圖(SEM);CoS2電極電化學(xué)表征(d)和穩(wěn)定性測(cè)試(e)���;(f)不同結(jié)構(gòu)的CoS2氫氣逸出示意圖
在燃料電池催化劑設(shè)計(jì)中���,納米結(jié)構(gòu)的優(yōu)化同樣十分重要。在長(zhǎng)時(shí)間的操作過(guò)程中���,ORR催化劑中除了自身活性衰減外���,還可能會(huì)因水淹而導(dǎo)致燃料電池快速失活。由于多孔通道被積水阻塞���,水淹將中斷活性位點(diǎn)的氧氣供應(yīng)���,導(dǎo)致淹沒(méi)區(qū)域的ORR終止。為了量化燃料電池中電催化劑的孔特性對(duì)其傳質(zhì)和抗水淹性能的影響���,Wang等為ORR催化劑設(shè)計(jì)了一種特殊的“撥浪鼓”狀工作電極���。雙級(jí)孔隙Pt/C催化劑具有較大的孔容和規(guī)整的孔道結(jié)構(gòu)���,其傳質(zhì)性能和抗水淹性能是工業(yè)催化劑的4倍。事實(shí)上���,不同類型的孔隙在ORR過(guò)程中具有特殊的作用���。在ORR過(guò)程中,中孔和大孔對(duì)傳質(zhì)過(guò)程中更重要���,而微孔有利于容納大多數(shù)催化位點(diǎn)。因此ORR催化劑需同時(shí)具有多級(jí)孔結(jié)構(gòu)���,以保證活性位點(diǎn)密度和傳質(zhì)效率���。
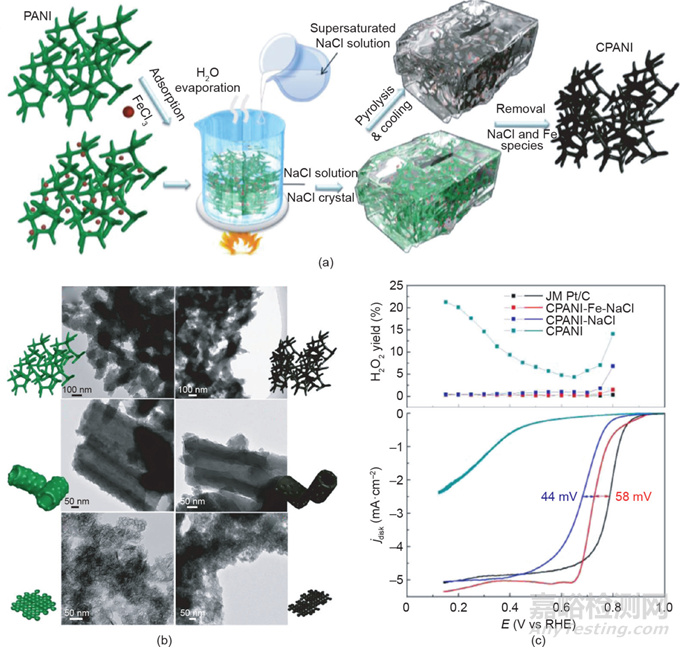
為構(gòu)建催化劑多級(jí)孔結(jié)構(gòu)���,以二氧化硅膠體���、有序介孔二氧化硅���、聚苯乙烯微球和其他一些氧化物為模板的犧牲模板法得到了廣泛研究和應(yīng)用。例如���,Liang等使用膠體二氧化硅為模板合成了比表面積高達(dá)1280 m2·g–1的氮摻雜碳催化劑,且該催化劑為具有介孔 / 微孔分布的多級(jí)孔結(jié)構(gòu)材料���。然而���,以上的犧牲模板法的模板去除步驟可能會(huì)十分耗時(shí)���,且該步驟通常需要使用強(qiáng)酸或強(qiáng)堿溶液,這對(duì)研究人員和環(huán)境可能帶來(lái)危害。為了避免這些缺點(diǎn)���,開(kāi)發(fā)了一種以NaCl重結(jié)晶為模板的形態(tài)學(xué)控制方法���。在這種方法中���,NaCl模板可以用熱水溶解而去除���,且NaCl模板可循環(huán)使用���。通過(guò)鹽重結(jié)晶,具有特殊納米結(jié)構(gòu)的聚苯胺(PANI)被封裝在NaCl晶體中���,然后在高溫下精確地轉(zhuǎn)化為碳納米材料(圖6)���。在高溫焙燒過(guò)程中,碳納米材料在NaCl晶體封閉的納米反應(yīng)器中氣化���,從而產(chǎn)生了大量的孔���。所制備的3D-Fe/N-C催化劑具有多孔隙、高活性位點(diǎn)利用率的特點(diǎn)���,對(duì)ORR具有良好的催化性能���。
圖6 (a)鹽重結(jié)晶法形貌固定示意圖;(b)所制的3D納米結(jié)構(gòu)聚苯胺(PANI)和對(duì)應(yīng)碳化產(chǎn)物的透射電鏡圖(TEM)���;(c)不同樣品的H2O2產(chǎn)出曲線和ORR曲線
(二)晶面工程
晶面調(diào)控是調(diào)節(jié)材料對(duì)給定反應(yīng)的催化性能的一種廣泛研究的方法���。由于催化反應(yīng)中間產(chǎn)物在催化劑不同晶面的吸附強(qiáng)度差異很大,催化材料的反應(yīng)活性與其暴露晶面高度相關(guān)���。晶面通常是用Miller指數(shù)表示的���。納米材料的暴露晶面與納米顆粒的形狀密切相關(guān)。一般來(lái)說(shuō)���,納米材料的晶面可分為低指數(shù)和高指數(shù)晶面類型���。低指數(shù)晶面是指Miller指數(shù)(hkl)三個(gè)組成部分之和較小的指數(shù)晶面���,而高指數(shù)晶面至少包含一個(gè)大于1的Miller指數(shù)。
低指數(shù)晶面納米材料反應(yīng)性的結(jié)構(gòu)敏感性在單晶Pt催化析氫反應(yīng)中已經(jīng)得到證實(shí)���。利用掃描隧道顯微鏡(STM���,圖7)證實(shí)了具有清晰表面的Pt(hkl)的表面形貌,并觀察到其活性程度在堿性溶液中遵循(110)>(100)>(111)的順序���。重要的是���,Pt的不同晶面的活性趨勢(shì)在堿性和酸性電解質(zhì)中有很大的不同。Markovic等重點(diǎn)研究了Pt(111)和Pt島修飾Pt(111)���,證實(shí)了這種pH效應(yīng)涉及HER中的結(jié)構(gòu)功能關(guān)系���。與原始Pt (111)表面的HER活性相比,Pt島修飾Pt(111)電極上的HER活性在堿性溶液中提高了5~6倍���,而在酸性電解液中的活性僅提高了1.5倍���。pH值對(duì)其活性的影響被證明是由于邊緣臺(tái)階位點(diǎn)解離水的特殊能力引起的���。此外,Pt(hkl)的活性順序與低配位Pt原子的密度一致���,這是由于低配位Pt原子加速了水的解離步驟。在非吸附性HClO4電解質(zhì)中���,單晶Pt不同晶面的ORR活性也觀察到類似的變化趨勢(shì)���。當(dāng)電解質(zhì)替代為H2SO4時(shí),Pt(100)比Pt(111)更為活躍���。這種活性差異是由于硫酸氫鈉陰離子在Pt(111)上的特殊吸附行為所致���。硫酸氫鈉陰離子在Pt(111)表面的吸附比在Pt(100)表面的吸附更強(qiáng),導(dǎo)致ORR的后續(xù)步驟受阻���。以上研究證實(shí)了各晶面的不同性質(zhì)對(duì)其催化性能有著顯著的影響���。在這項(xiàng)工作之后,許多研究集中在開(kāi)發(fā)一種在催化表面上可控構(gòu)建特定晶面的方法���,以應(yīng)用于理想的單晶金屬及實(shí)用的納米材料���。El Sayed等首次發(fā)現(xiàn)了鉑納米晶體的晶面控制合成���,制備得到富含(100)晶面的納米立方體、(111)晶面的納米四面體以及同時(shí)具有(111)和(100)晶面的納米球���。Sun等報(bào)道了一種高溫有機(jī)相法用于合成單分散(100)端Pt納米立方體���。晶面控制的Pt納米立方體在酸性電解液中的ORR活性是工業(yè)Pt催化劑活性的兩倍以上。

圖7 (a)Pt不同晶面的HER/HOR���;(b)~(d)Pt不同晶面的STM(插圖為其對(duì)應(yīng)的結(jié)構(gòu)模型)���;(e)制備的Pt (111)和島狀Pt/Pt (111)表面的 STM圖;(g)兩個(gè)Pt晶面的HER活性曲線圖
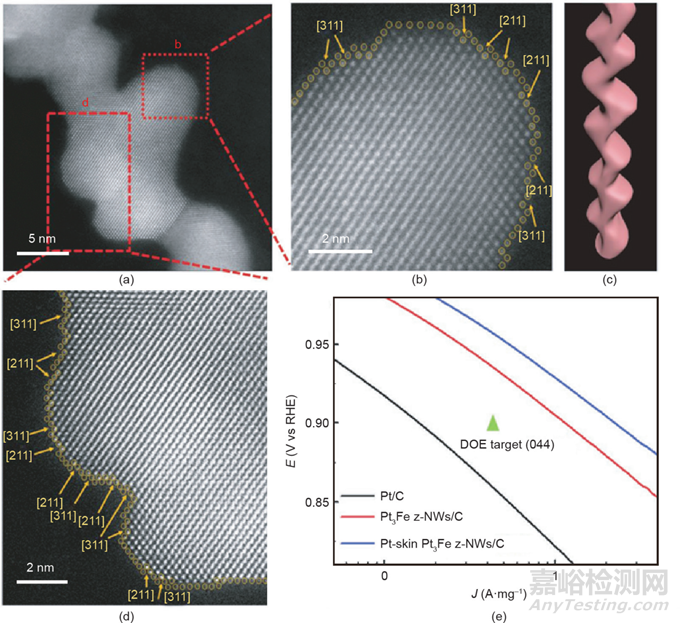
由于低配位原子���、臺(tái)階���、邊緣和扭結(jié)的密度更高,具有高指數(shù)晶面的金屬及化合物與典型的低指數(shù)材料相比通常具有更高的反應(yīng)性���。然而���,由于其較高的表面能���,高指數(shù)晶面是熱力學(xué)上不穩(wěn)定的。因此���,制備高指數(shù)晶面納米材料是一項(xiàng)艱巨的任務(wù)���。近年來(lái)���,為了提高催化活性���,人們開(kāi)發(fā)了多種方法來(lái)合成具有高指數(shù)晶面的納米材料。Xia等開(kāi)發(fā)了一種在水溶液中還原的簡(jiǎn)單路線���,以制備由(510)���、(720)和(830)高指數(shù)晶面包圍的Pt凹面納米立方體(c-NCs)。利用甘氨酸控制H2PtCl6的還原動(dòng)力學(xué)也可以制備得到Pt c-NCs���。Sun等采用電化學(xué)方法合成了具有(730)���、 (210)和(520)晶面的四面體(THH)Pt納米晶���。除了單金屬材料外,多金屬高指數(shù)晶面納米晶體的合成方法也得到了開(kāi)發(fā)���。如圖8所示���,Guo等報(bào)道了一類具有穩(wěn)定高指數(shù)晶面和表面富Pt結(jié)構(gòu)的新型Pt3Fe鋸齒狀納米線(z-NWs)。這些獨(dú)特的結(jié)構(gòu)特征賦予了Pt3Fe z-NWs優(yōu)異的ORR活性���,其在0.9 V(vs. RHE)下的質(zhì)量活性和比活性分別為2.11 A·mg–1和4.34 mA·cm–2���。
圖8 (a)Pt-skin Pt3Fe z-NWs的高角度環(huán)形暗場(chǎng)(HAADF)-TEM圖;(b)���、(d)圖(a)中紅色方塊區(qū)域的放大圖���;(c)鋸齒狀納米線的結(jié)構(gòu)示意圖;(e)商業(yè)化Pt/C、Pt3Fe z-NWs/C和Pt-skin Pt3Fe z-NWs/C的ORR質(zhì)量活性圖
為了降低催化劑成本���,設(shè)計(jì)和制備具有不同晶面的非貴金屬納米材料已成為晶面工程的研究熱點(diǎn)���。Su等研究了NiO晶體的生長(zhǎng)機(jī)制,發(fā)現(xiàn)NiO晶體的表面能遵循(100)< (113)<(101)≈(110)的順序���。Han等提供了一種無(wú)模板水熱方法���,用于可控地制備具有(001)、(112)和(001)+(111)晶面的Co3O4納米立方體(NC)���、納米八面體(NTO)和納米多面體(NP)���。不同的晶面賦予Co3O4納米晶表面的Co2+和Co3+活性位以不同原子結(jié)構(gòu)���。在含有豐富Co3+位點(diǎn)的還原氧化石墨烯(rGO)上���,(112)晶面覆蓋的Co3O4納米顆粒對(duì)OER和ORR均表現(xiàn)出良好的活性。除了金屬氧化物外���,許多具有特殊晶面的金屬化合物也相繼被報(bào)道���。Feng等合成了具有穩(wěn)定(210)面的Ni3S2納米片陣列���,展現(xiàn)了高效穩(wěn)定的HER和OER電催化性能。Pan等制得了具有不同晶體結(jié)構(gòu)的花狀磷化鎳(Ni5P4和Ni2P)���,并證明其優(yōu)異的催化活性歸因于具有高能(001)晶面的多級(jí)結(jié)構(gòu)���。
(三)晶相工程
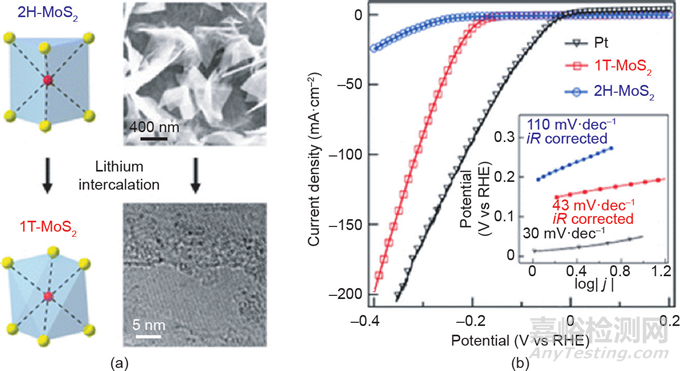
除了調(diào)整納米晶體的暴露晶面外,調(diào)節(jié)原子尺度的排列(即晶相的轉(zhuǎn)變)也會(huì)使催化劑的物理和化學(xué)性質(zhì)發(fā)生根本性的變化���,從而影響催化劑的本征活性���。具有獨(dú)特多晶相的過(guò)渡金屬二硫化物是被廣泛研究的典型案例。在這些多晶型中���,亞穩(wěn)態(tài)1T相由于其金屬性的性質(zhì)引起了極大的研究興趣���。Jin等用鋰插層法從半導(dǎo)體2H-MoS2合成了金屬性1T-MoS2納米片。與相應(yīng)的2H晶相相比���,1T-MoS2在電催化HER方面表現(xiàn)出顯著改善的性能(圖9)���。類似地���,Jin等通過(guò)更簡(jiǎn)單的微波輔助插層法合成了金屬性1T二硫化鎢(1T-WS2)。晶相工程賦予了1T-WS2更優(yōu)異的導(dǎo)電性和更密集的活性位點(diǎn)���,增強(qiáng)了其析氫催化活性���。1T催化劑不僅可以通過(guò)改變?cè)优帕蝎@得獨(dú)特的性質(zhì),而且其活性中心也可能不同于傳統(tǒng)的2H結(jié)構(gòu)相���。Voiry等通過(guò)去除化學(xué)剝落的MoS2納米片表面的剩余負(fù)電荷���,獲得了具有優(yōu)異活性的高導(dǎo)電性1T-MoS2納米片。有趣的是���,1T-MoS2和2H-MoS2部分氧化后,其活性變化形成鮮明對(duì)比���。邊緣氧化后1T-MoS2的HER活性幾乎沒(méi)有變化���,但2H-MoS2的活性卻嚴(yán)重下降���。眾所周知,通常2H-MoS2晶體的邊緣是其主要的活性中心���。部分氧化的1T-MoS2和2H-MoS2在HER活性上的顯著差異表明���,1T-MoS2催化的主要活性中心不是納米片的邊緣,而是納米片的基面���。
圖9 (a)經(jīng)過(guò)鋰插層后由半導(dǎo)體2H-MX2到金屬態(tài)1T-MX2的相轉(zhuǎn)變���;(b)兩種形態(tài)MoS2的HER極化曲線
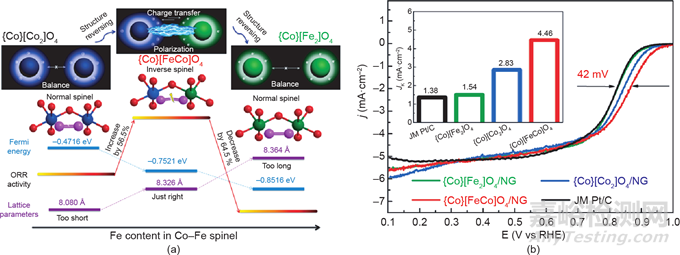
金屬氧化物的催化性能也受其晶相影響。Wu等發(fā)現(xiàn)反轉(zhuǎn)尖晶石晶體結(jié)構(gòu)對(duì)尖晶石的ORR催化活性有很大影響(圖10)���。通過(guò)調(diào)整鐵(Fe)的含量���,Co–Fe基晶體的尖晶石結(jié)構(gòu)可以從正常結(jié)構(gòu)變?yōu)榉唇Y(jié)構(gòu),后再變回正常結(jié)構(gòu)���。電化學(xué)結(jié)果表明���,具有反尖晶石結(jié)構(gòu)的{Co}[FeCo]O4/NG具有最好的ORR活性���。DFT結(jié)果進(jìn)一步揭示了反尖晶石結(jié)構(gòu)的{Co}[FeCo]O4/NG的高ORR活性是由于八面體位置的Fe和Co原子的異化效應(yīng)引起的氧吸附能的改變和氧-氧鍵的拉長(zhǎng)所致。此外���,晶相對(duì)ORR反應(yīng)途徑的影響也有所報(bào)道���。Karunagaran等合成了四種不同晶相的氧化鐵納米顆粒負(fù)載于三維rGO氣凝膠,并測(cè)定了它們催化ORR的電化學(xué)性能和電子轉(zhuǎn)移���。結(jié)果表明���,在高電位(0.70 V)下,四種催化劑均通過(guò)雙電子途徑催化ORR���。當(dāng)電位降低到0.20 V時(shí)���,ORR在含磁鐵礦、磁赤鐵礦和針鐵礦的rGO復(fù)合材料通過(guò)四電子轉(zhuǎn)移動(dòng)力學(xué)進(jìn)行���,而在含赤鐵礦的復(fù)合材料則通過(guò)兩電子轉(zhuǎn)移動(dòng)力學(xué)進(jìn)行���。
圖10 (a)結(jié)構(gòu)轉(zhuǎn)換和ORR活性之間的關(guān)系;(b){Co}[Fe2]O4/NG���、{Co}[Co2]O4/NG���、{Co}[FeCo]O4/NG和Pt/C的ORR活性
過(guò)渡金屬化合物的Jahn-Teller效應(yīng)引起的構(gòu)型畸變與其電催化性能的關(guān)系也有大量研究。Liu等通過(guò)對(duì)Co3S4/TETA雜化前驅(qū)體的超聲剝離得到原子層厚度的Co3S4納米片(CSATNs)���,并觀察到CSATNs產(chǎn)物存在明顯的結(jié)構(gòu)畸變���。CSATNs的結(jié)構(gòu)畸變引起電子結(jié)構(gòu)的改變。與塊體樣品相比[圖11(a)���、(b)]���,CSATNs的譜圖向低磁場(chǎng)的偏移意味著其八面體中心(t2g4eg2)中Co3+的自旋狀態(tài)從低自旋調(diào)整到高自旋���。高角度環(huán)形暗場(chǎng)(HAADF)圖像顯示���,八面體配位陽(yáng)離子僅暴露在平面中���,進(jìn)一步揭示了Jahn-Teller拉伸的存在[圖11(c)~(f)]���。由于原子和電子結(jié)構(gòu)的協(xié)同調(diào)整���,CSATNs具有明顯的增強(qiáng)OER性能���。事實(shí)上���,Jahn-Teller效應(yīng)是由于簡(jiǎn)并軌道(t2g或eg)中心離子的電子分布不均勻造成的。因此���,eg軌道上電子的填充態(tài)對(duì)過(guò)渡金屬化合物的催化性能有著重要的影響。Shao-Horn等進(jìn)一步發(fā)現(xiàn)了鈣鈦礦基氧化物(ABO3)中B離子的eg軌道的填充狀態(tài)與ORR活性之間的火山關(guān)系[圖11(g)]���。當(dāng)鈣鈦礦型氧化物eg軌道上只有一個(gè)電子填充時(shí)(定義為eg≈1)���,其具有最高的ORR活性,因?yàn)榇藭r(shí)O2可以以最佳結(jié)合能吸附在B位���。雖然尖晶石的ORR活性位不是四面體位而是八面體位���,但eg占有率理論也可以進(jìn)一步擴(kuò)展應(yīng)用于尖晶石氧化物的活性描述[圖11(h)]���。
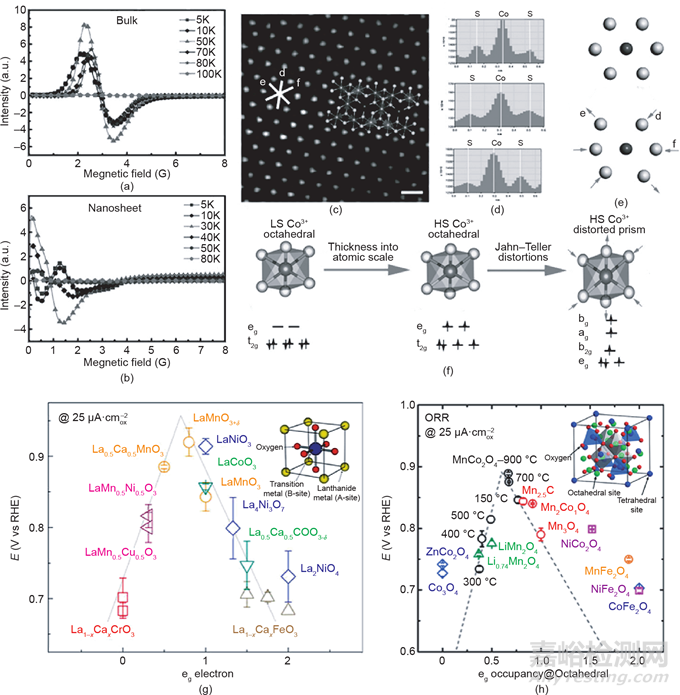
圖11 (a)、(b)塊狀和Co3S4納米片的電子順磁共振圖(EPR)���;CSATNs的HAADF(c)和強(qiáng)度線(d);Jahn-Teller變形(e)和結(jié)構(gòu)轉(zhuǎn)變(f)圖示���;(g)鈣鈦礦氧化物的ORR活性對(duì)eg電子的函數(shù)圖���;(h)在尖晶石氧化物的ORR活性中位于八面體位點(diǎn)的活性元素eg的作用
(四)非晶化
通過(guò)非晶化來(lái)調(diào)節(jié)原子尺度的排列,是提高材料催化性能另一個(gè)研究熱點(diǎn)���。非晶相的短程原子排列有利于提高活性中心的密度。早在1995年���,Weber等研究了非晶態(tài)化合物MoS3的結(jié)構(gòu)單元���,發(fā)現(xiàn)所有鉬都處于Mo4+氧化狀態(tài),而硫原子則以兩種不同的配位形式存在:S2–和S22–���。Hu等證實(shí)非晶態(tài)MoS2在催化HER方面更為活躍���。非晶態(tài)MoSx薄膜表面極為粗糙���,硫元素含量豐富���,催化活性區(qū)大,活性中心密集���。Benck等進(jìn)一步揭示了非晶態(tài)硫化鉬的HER活性的增加是非晶態(tài)結(jié)構(gòu)和納米結(jié)構(gòu)所引起的大量活性位點(diǎn)的作用。同時(shí)���,Li等從組成和結(jié)晶度方面系統(tǒng)地研究了非晶態(tài)MoS2催化活性的來(lái)源���。有趣的是,實(shí)驗(yàn)結(jié)果表明結(jié)晶度是決定催化性能的關(guān)鍵���,而組成并不特別重要���。
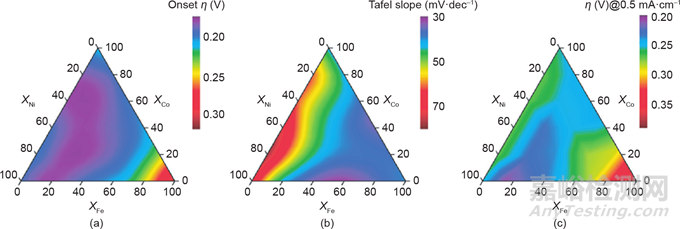
除了HER催化,Smith等基于對(duì)鐵���、鎳和鈷混合金屬氧化物的研究���,證明非晶態(tài)材料比晶體材料具有更佳的OER催化活性���。對(duì)于非晶態(tài)結(jié)構(gòu),金屬在整體材料中的分布是均勻的���,且其成分可以精確控制。以最佳元素含量配比制備的a-Fe100-y-zCoyNizOx 催化劑具有優(yōu)異的催化性能���,甚至可以與工業(yè)貴金屬氧化物催化劑相媲美���。基于非晶態(tài)材料的組成可控的特點(diǎn)���,可以進(jìn)一步研究金屬組成以及非晶化對(duì)電催化性能的影響���。Smith等制備了21個(gè)復(fù)合金屬氧化物薄膜用于電催化水氧化,并測(cè)量了每個(gè)樣品中Fe���、Co和Ni的準(zhǔn)確化學(xué)計(jì)量濃度。電化學(xué)測(cè)量證實(shí)鐵含量對(duì)降低Tafel斜率很重要���,而鈷或鎳有利于降低過(guò)電位(圖12)���。由于無(wú)定形態(tài)催化劑優(yōu)異的催化性能���,其規(guī)?��;a(chǎn)方法的研究十分重要。Kuai等提出了一種噴霧輔助的方法���,通過(guò)這種方法可以可持續(xù)地獲得非晶混合金屬氧化物���,非常適合工業(yè)應(yīng)用。所制備的Fe6Ni10Ox在電化學(xué)OER中驅(qū)動(dòng)10 mA·cm–2時(shí)表現(xiàn)出0.286 V的低過(guò)電位���,且Tafel斜率僅為48 mV·decade–1���,優(yōu)于所有研究的Fe-Ni-Ox 催化劑的催化性能���。
圖12 不同金屬成分的無(wú)定形金屬氧化物薄膜活性參數(shù)的等高線���。
(a)起始電位���;(b)塔菲爾曲線;(c)j=0.5 mA·cm–2時(shí)的過(guò)電位
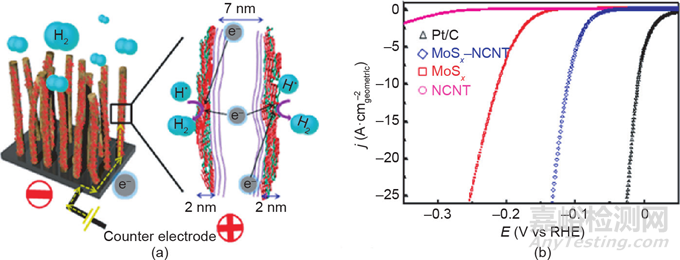
雖然非晶態(tài)工程可以大大提高催化劑的活性位密度���,但由于晶體結(jié)構(gòu)的短程無(wú)序���,非晶態(tài)材料的導(dǎo)電性會(huì)降低���。將低導(dǎo)電非晶態(tài)材料與高導(dǎo)電材料復(fù)合是保證非晶催化劑優(yōu)良電催化性能的有效途徑���。Lee等合成了以低成本、高導(dǎo)電性的Ketjenback(KB)碳負(fù)載非晶MnOx納米線作為高效ORR電極���,大大加速了電催化過(guò)程中的電子轉(zhuǎn)移���。許多其他非晶態(tài)/導(dǎo)電復(fù)合材料,如非晶態(tài)MoSx/碳復(fù)合催化劑���、非晶態(tài)MoSx/聚吡咯共聚物薄膜(PPy/MoSx)和非晶態(tài)MoSx/氮摻雜碳納米管催化劑(NCNT)也被報(bào)道。這些復(fù)合材料中的高導(dǎo)電骨架可以克服非晶態(tài)催化劑的低導(dǎo)電性引起的障礙���,從而顯著提高催化活性(圖13)���。多孔金屬納米結(jié)構(gòu)���,如鎳泡沫和納米多孔金���,也被用作支持非晶態(tài)MoSx催化劑的導(dǎo)電基底���,以顯著增強(qiáng)其HER活性。
圖13 (a)MoSx/NCNT森林狀雜化催化劑的HER示意圖���;(b)不同樣品的HER活性曲線
(五)缺陷工程
缺陷普遍存在于納米材料中。人們已經(jīng)認(rèn)識(shí)到���,缺陷催化劑表面總是比無(wú)缺陷催化劑表現(xiàn)出更高的反應(yīng)活性。因此���,缺陷工程逐漸發(fā)展成為調(diào)整納米材料電子和表面性質(zhì)的有效方法���。
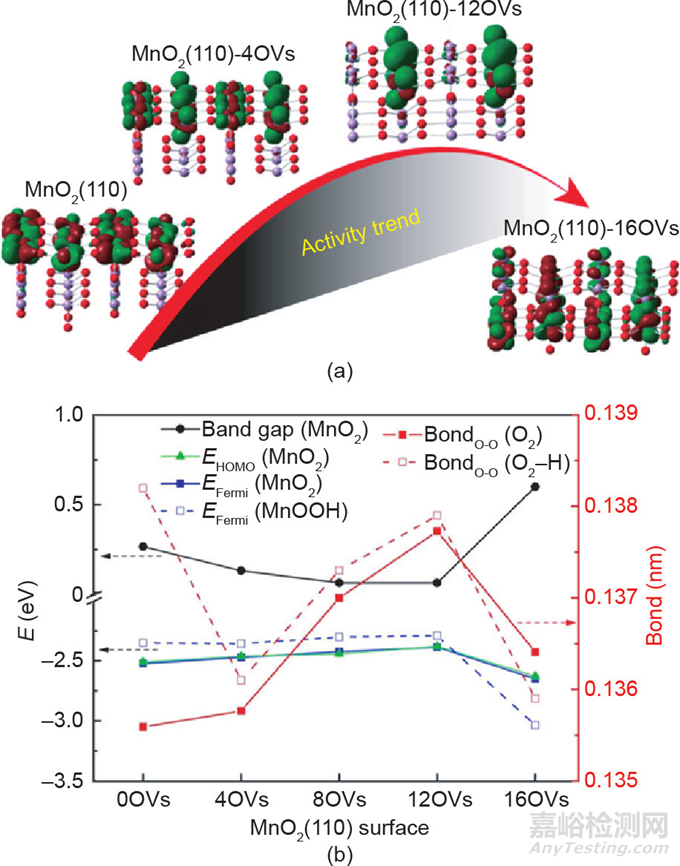
Cheng等通過(guò)在環(huán)境條件下水溶液中還原非晶態(tài)二氧化錳,合成了四方或立方MxMn3–xO4尖晶石���。納米MxMn3–xO4由于其高活性面積和豐富的缺陷���,對(duì)ORR和OER都具有相當(dāng)高的催化活性���。同樣���,Qiao等合成了富氧空位(OV)缺陷的介孔MnCo2O4材料���,發(fā)現(xiàn)其穩(wěn)定性和耐甲醇性能甚至超過(guò)了Pt/C催化劑���。為了深入了解缺陷對(duì)催化性能的影響,Li等應(yīng)用DFT+U計(jì)算研究了OV濃度對(duì)β-MnO2催化劑電子結(jié)構(gòu)及其ORR的催化性能的影響���。如圖14所示,電子結(jié)構(gòu)與OV濃度的曲線關(guān)系表明���,OV濃度可以調(diào)節(jié)β-MnO2的電導(dǎo)率和ORR催化活性���。適當(dāng)濃度的OVs將大大提高M(jìn)nO2的電導(dǎo)率���,而過(guò)量的OVs將阻礙ORR過(guò)程���。缺陷工程也可用于提高納米材料催化活性位點(diǎn)的密度。Xie等設(shè)計(jì)了一種高濃度前驅(qū)體���、不同硫脲用量的反應(yīng)���,實(shí)現(xiàn)了對(duì)所制備超薄MoS2納米片缺陷的可控調(diào)制。由于其富含缺陷的結(jié)構(gòu)���,MoS2納米片表面形成了許多細(xì)小的裂紋���,使得其活性位點(diǎn)數(shù)量是無(wú)缺陷MoS2的13倍���。
圖14 (a)不同OV濃度下MnO2的活性趨勢(shì)���;(b)DFT計(jì)算各性質(zhì)結(jié)果隨OV濃度的改變
與金屬化合物中的空位類似���,碳基電催化劑中的固有缺陷是普遍存在的���,但長(zhǎng)期以來(lái)卻一直被忽略���。通常���,碳材料經(jīng)過(guò)雜原子摻雜后容易形成缺陷,成為有利于電催化的活性中心���。然而���,雜原子摻雜碳基材料的電催化活性主要?dú)w因于雜原子摻雜的誘導(dǎo)效應(yīng)���。隨著時(shí)間的推移���,一些研究發(fā)現(xiàn),具有本征缺陷的碳電催化劑的催化活性甚至優(yōu)于雜原子摻雜碳材料���。
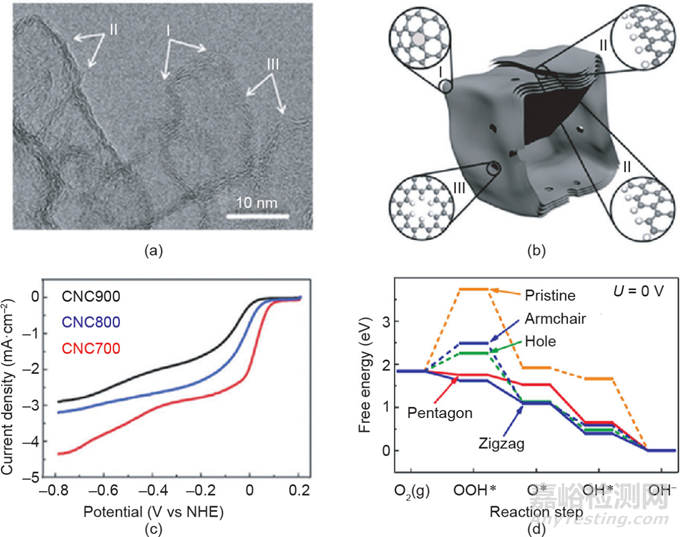
Hu等發(fā)現(xiàn)缺陷碳納米籠(CNC)具有比硼摻雜碳納米管更高的ORR活性。Hu等成功地合成了缺陷豐富的CNC���,該材料具有許多典型的缺陷位置���,但沒(méi)有任何摻雜劑[圖15(a)]。電化學(xué)結(jié)果表明,缺陷密度最高的CNC材料表現(xiàn)出最好的ORR電化學(xué)活性[圖15(c)]���。DFT結(jié)果進(jìn)一步表明,這些缺陷材料的高ORR活性可歸因于五邊形和鋸齒形邊緣缺陷[圖15(d)]���。Yao等利用第一性原理計(jì)算���,預(yù)測(cè)了石墨烯上585種缺陷的ORR活性甚至超過(guò)了氮摻雜位點(diǎn)���,并通過(guò)實(shí)驗(yàn)研究為這一理論預(yù)測(cè)提供有力支持���。考慮到缺陷機(jī)制���,Yao等通過(guò)在950 ℃下碳化Zn-MOF制備了一種無(wú)元素?fù)诫s的多孔碳(PC)材料。通過(guò)去除鋅原子���,可以在PC催化劑上形成缺陷���,使PC催化劑不僅具有優(yōu)異的ORR活性,而且具有與商業(yè)Pt/C催化劑相當(dāng)?shù)姆€(wěn)定性。此外���,除ORR過(guò)程外���,在從氫-水轉(zhuǎn)化系統(tǒng)中其他三種電化學(xué)反應(yīng)(即HOR���、OER和HER)的電催化活性被證明對(duì)石墨烯中的缺陷類型特別敏感。
圖15 CNC700的高倍透射電鏡圖(a)和結(jié)構(gòu)示意圖(b)���;(c)CNC樣品的旋轉(zhuǎn)圓盤(pán)電極(RDE)ORR活性曲線;(d)不同缺陷的ORR步驟的自由能圖
(六)原子摻雜
原子摻雜是調(diào)節(jié)催化材料性能最常用的策略���。通過(guò)合理地將一個(gè)或多個(gè)金屬或非金屬元素引入材料的晶格中���,可以調(diào)節(jié)原材料的電子結(jié)構(gòu)���,從而有效地提高材料的催化性能���。以MoS2為例,多種金屬元素如Ni、Co���、Fe���、V���、Li和Cu被成功摻雜到其晶體結(jié)構(gòu)中���,有效改善了其物理和化學(xué)特性。在這些摻雜的金屬元素中���,Ni和Co傾向于摻雜在MoS2中S元素的附近,這將降低S邊緣的氫吸附能���,增加MoS2材料中活性位點(diǎn)的密度���。與Ni和Co的摻雜不同,V摻雜不能增加活性位點(diǎn)的數(shù)量���,但會(huì)增強(qiáng)MoS2的導(dǎo)電性���。
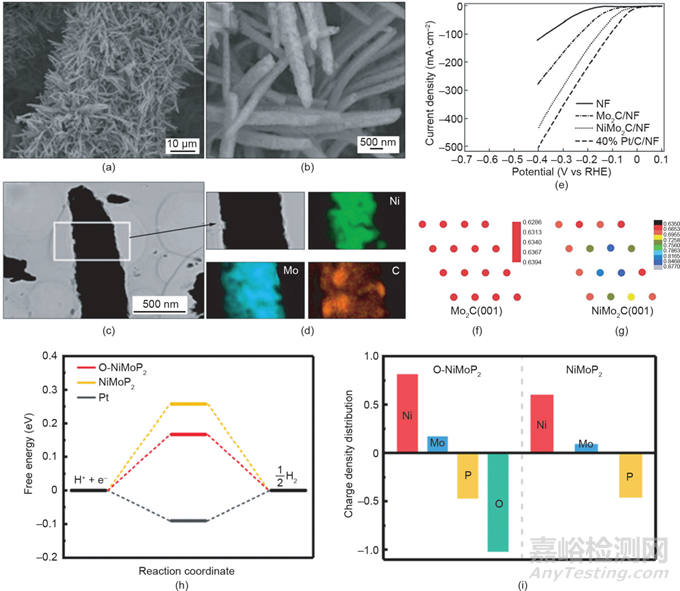
Xiong等通過(guò)結(jié)合實(shí)驗(yàn)和理論計(jì)算,探討了碳化鉬中鎳的摻雜對(duì)其表面電子結(jié)構(gòu)的影響及與其催化性能之間的關(guān)系���。如圖16(a)~(d)所示,通過(guò)水熱和碳化處理���,一維NiMo2C納米線陣列直接構(gòu)建在導(dǎo)電泡沫鎳上(NiMo2C/NF)。與Mo2C和Ni催化劑相比���,這種無(wú)黏結(jié)劑的NiMo2C/NF集成電極顯示出更優(yōu)異的催化活性[圖16(e)]���。DFT計(jì)算結(jié)果表明,Ni與Mo2C晶格的結(jié)合改變了催化劑的電荷分布���,產(chǎn)生了Ni與Mo2C的協(xié)同效應(yīng),從而降低了氫結(jié)合能[圖16(f)���、(g)]���。除了金屬元素���,非金屬元素?fù)诫s的研究也非?��;钴S���。Xie等成功合成了氧摻雜MoS2超薄納米片,可以協(xié)同調(diào)控其活性位點(diǎn)和導(dǎo)電性���。DFT計(jì)算結(jié)果顯示,含氧MoS2的微分結(jié)合能更小���,其驅(qū)動(dòng)HER過(guò)程的能壘更低���。Zhang等通過(guò)部分磷化金屬氧化物前驅(qū)體,構(gòu)建了氧摻雜NiMoP2(O-NiMoP2)���。如圖16(h)~(i)所示,氧的摻入優(yōu)化了NiMoP2表面的氫吸附能���,其ΔGH*比未摻雜樣品更接近于零���。此外���,O-NiMoP2中的Ni和Mo帶有更多的正電荷���,這有利于水分子的吸附和活化���,極大加速了水在堿性介質(zhì)中的解離���。
圖16 (a)~(d)NiMo2C電極的掃描電鏡���、透射電鏡圖和元素能譜圖���;(e)NiMo2C電極的電化學(xué)析氫活性曲線;Mo2C(001)(f)和NiMo2C(001)(g)的Bader電荷分布圖���;不同NiMo基樣品的氫自由能(h)和電荷密度分布(i)圖
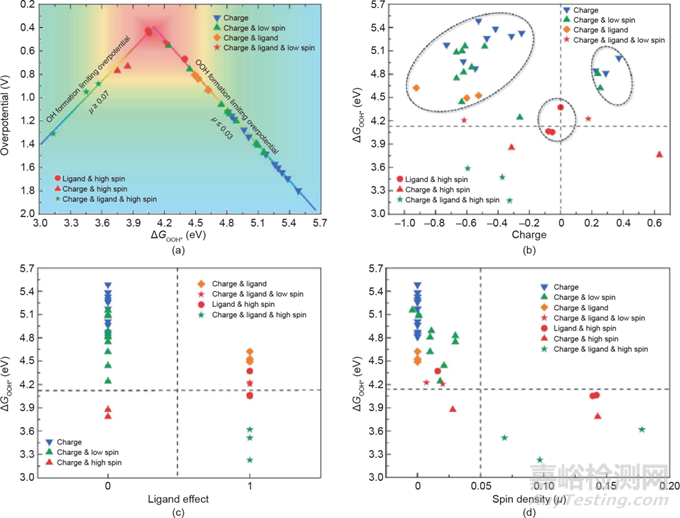
除了金屬化合物,碳基材料也被廣泛用作摻雜目標(biāo)���,極大地?cái)U(kuò)大了催化劑的研究范圍。2013年���,Li等報(bào)道了一種磷摻雜石墨烯���,其ORR催化性能與商業(yè)用Pt/C相當(dāng)。隨后���,Li等進(jìn)一步制備了氮磷雙摻雜石墨烯材料作為ORR和OER的電催化劑���,其催化活性超過(guò)了商業(yè)Pt/C催化劑。為了揭示雜原子摻雜碳高活性的起因���,Yang等對(duì)一系列不同雜原子摻雜的石墨烯進(jìn)行了全面的DFT計(jì)算���。DFT結(jié)果表明���,摻雜碳催化劑存在三重效應(yīng),即電荷���、自旋密度和配體效應(yīng)決定了摻雜碳催化劑的本征催化活性及 ORR機(jī)制(圖17)���。當(dāng)碳材料被單一雜原子摻雜時(shí)���,摻雜原子周圍的碳位只能被單一效應(yīng)激活���。這導(dǎo)致ORR 通過(guò)結(jié)合機(jī)制進(jìn)行,并且存在不低于0.44 V本征過(guò)電位限制���。當(dāng)碳材料被金屬或雙雜原子摻雜時(shí),雙碳位可以被三重效應(yīng)激活���,其ORR遵循離解機(jī)制���。此時(shí)���,結(jié)合機(jī)制的活性限制將不再有效���,從而ORR活性可得到有效增強(qiáng)���。Huang等合成了金屬和非金屬元素共摻雜的石墨烯,并揭示了氮摻雜石墨烯中的氮結(jié)構(gòu)和微量金屬原子對(duì)HER催化的作用���。該研究發(fā)現(xiàn)���,季銨態(tài)氮是氮摻雜石墨烯中三種摻雜氮類型中最活躍的位點(diǎn)。而當(dāng)摻雜微量鈷原子時(shí)���,平面氮的活性最強(qiáng);當(dāng)微量鈷原子被鎳取代時(shí)���,平面氮的活性則被抑制���。
圖17 (a)碳活性位點(diǎn)的ORR過(guò)電位對(duì)ΔGOOH*函數(shù)圖���;ΔGOOH*對(duì)電荷效應(yīng)(b)���、配體效應(yīng)(c)和自旋密度關(guān)系(d)圖
(七)界面工程
雜化納米材料擁有一個(gè)位于兩個(gè)組分的邊界的界面。對(duì)于多相催化劑來(lái)說(shuō)���,具有適當(dāng)?shù)慕缑娼Y(jié)構(gòu)是極其重要的���,因?yàn)榻缑鎱^(qū)域總是呈現(xiàn)出獨(dú)特的物理和化學(xué)性質(zhì)���。這些獨(dú)特的性質(zhì)可以促進(jìn)材料結(jié)合���、轉(zhuǎn)化和運(yùn)輸表面物種的能力���,極大促進(jìn)了發(fā)生在其表面的催化反應(yīng)。近年來(lái)���,有大量的研究通過(guò)界面工程設(shè)計(jì)與合成催化劑用于氫-水轉(zhuǎn)化���。
一般來(lái)說(shuō)���,根據(jù)組分的相對(duì)位置,雜化材料可分為支撐結(jié)構(gòu)���、異質(zhì)結(jié)構(gòu)或核殼結(jié)構(gòu)���。支撐結(jié)構(gòu)的特點(diǎn)是支撐組分比其他組分大得多���,而異質(zhì)結(jié)構(gòu)材料中的組分尺寸相似���。在核殼結(jié)構(gòu)中���,一個(gè)組分被另一個(gè)組分覆蓋,在兩個(gè)組分之間的邊界處存在界面���。這三種具有不同界面結(jié)構(gòu)的雜化材料是近年來(lái)研究金屬���、金屬氧化物���、非氧化物等組裝的典型結(jié)構(gòu)���。例如���,F(xiàn)eng等通過(guò)控制鎳原子在煅燒過(guò)程中向外擴(kuò)散���,合成了一種MoNi4固定在MoO2長(zhǎng)方體上的支撐結(jié)構(gòu)電催化劑(MoNi4/MoO2@Ni)���,在堿性溶液中表現(xiàn)出優(yōu)異的HER活性[圖18(a)���、(b)]���。Xue等通過(guò)將金屬鈷逐漸磷化為CoP制備了異質(zhì)結(jié)構(gòu)Co/CoP納米顆粒。通過(guò)改變NaH2PO2和Co元素的比重���,異質(zhì)Co/CoP納米顆粒中的CoP含量能夠可控調(diào)節(jié)���,進(jìn)而改變Co/CoP催化劑的界面區(qū)[圖18(c)~(f)]���。如圖18(g)~(h)所示���,Li等通過(guò)對(duì)雙金屬(Ni,Co)有機(jī)骨架進(jìn)行碳化還原和可控氧化煅燒���,制備了由碳限域NiCo@NiCoO2納米顆粒組成的多孔納米陣列(NiCo@NiCoO2/C PMRAs)。所制得的NiCo@NiCoO2/C PMRAs包含了催化OER所需的幾種特性���,包括大的表面積、良好的導(dǎo)電性和豐富的電催化活性位點(diǎn)。
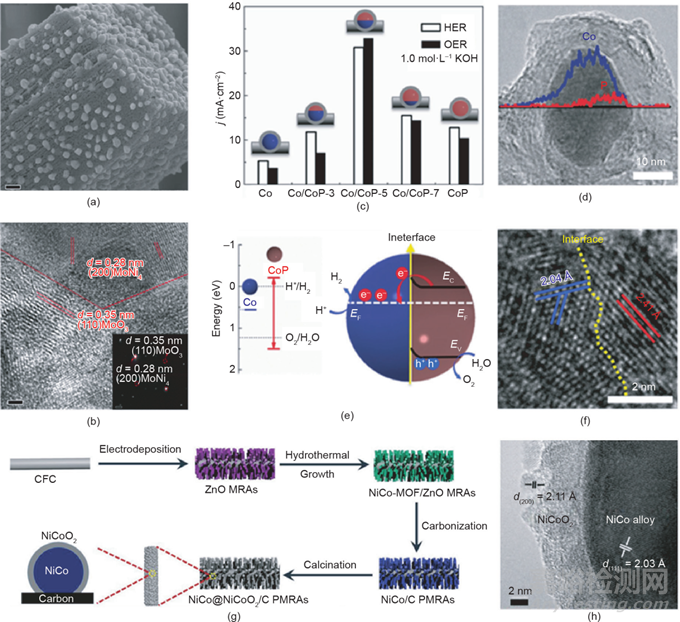
圖18 (a)���、(b)MoNi4/MoO2@Ni的典型SEM圖���;(c)制備樣品的電流密度���;(d)Co/CoP-5納米顆粒的組成曲線;(e)金屬Co���、CoP和基 Co/CoP的Mott-Schottky接觸的電子結(jié)構(gòu)���;(f)Co/CoP-5納米顆粒的HRTEM圖���;NiCo@NiCoO2/C PMRAs的制備過(guò)程(g)和HRTEM圖(h)
為了更合理地設(shè)計(jì)催化劑���,對(duì)具有豐富界面的雜化材料的催化性能提升原因也進(jìn)行了深入研究���。其中���,雜化材料中界面位點(diǎn)的電子結(jié)構(gòu)調(diào)控已被充分證實(shí)���。Yu等用X射線吸收近邊光譜(XANES)觀察到了Ni(OH)2/Pt催化劑的電子轉(zhuǎn)移���。在α-和β-Ni(OH)2/Pt電極上Ni的前邊緣和主吸收邊緣均向低能方向移動(dòng)���,說(shuō)明電子從Pt基底向氫氧化物轉(zhuǎn)移。
Hu等揭示了Pt/CoS2雜化體系中CoS2和Pt在水電解反應(yīng)中的強(qiáng)金屬載體相互作用(SMSI)。DFT計(jì)算預(yù)測(cè)了Pt的d帶結(jié)構(gòu)向下移動(dòng)���,這也進(jìn)一步被X射線光電子能譜(XPS)和X射線吸收精細(xì)結(jié)構(gòu)(XAFS)所證實(shí)���。盡管電子轉(zhuǎn)移在金屬/金屬?gòu)?fù)合催化劑中已經(jīng)得到了很好的證明���,但其高催化活性的潛在機(jī)制尚不明確���。為了揭開(kāi)這個(gè)謎團(tuán)���,Peng等研究了金屬/金屬氧化物界面的化學(xué)性質(zhì),發(fā)現(xiàn)了一種界面誘導(dǎo)的協(xié)同效應(yīng)——“煙囪效應(yīng)”[圖19(a)���、(b)]���。DFT計(jì)算結(jié)果表明[圖19(c)���、(d)]���,界面附近的位點(diǎn)對(duì)H2O*和OH*物種不吸附,而只選擇性地吸附H*���,這有效地避免了H2O*和OH*對(duì)活性位點(diǎn)的毒害作用���。同時(shí)���,界面上的活性位上H*反應(yīng)物種的ΔGH*接近于0���,說(shuō)明其對(duì)H*反應(yīng)物種的吸附和解吸能力良好。結(jié)合以上特點(diǎn)���,析氫反應(yīng)在金屬氧化物 / 金屬催化劑界面處連續(xù)進(jìn)行���,形成了類似于連續(xù)產(chǎn)氫的煙囪���。此外,金屬氧化物 / 金屬?gòu)?fù)合材料的HER活性與界面金屬原子之間呈正相關(guān)[圖19(e)],說(shuō)明可以通過(guò)增加界面活性位點(diǎn)的數(shù)量從而加速析氫過(guò)程���。除金屬/金屬?gòu)?fù)合催化劑外���,Peng等還通過(guò)制備單金屬NiONi3S2異質(zhì)節(jié)納米片進(jìn)一步研究了金屬/金屬?gòu)?fù)合催化劑性能提高的根源。NiO-Ni3S2界面處Ni–S鍵向Ni–O鍵的電子轉(zhuǎn)移導(dǎo)致了其比基準(zhǔn)Pt/C和RuO2催化劑具有更好的電解水活性[圖19(g)]。DFT計(jì)算結(jié)果表明���,NiO-Ni3S2界面上氫(或含氧)中間體的活化勢(shì)壘顯著降低���,說(shuō)明界面活性位點(diǎn)具有優(yōu)異的OER和HER本征活性[圖19(h)���、(i)]���。
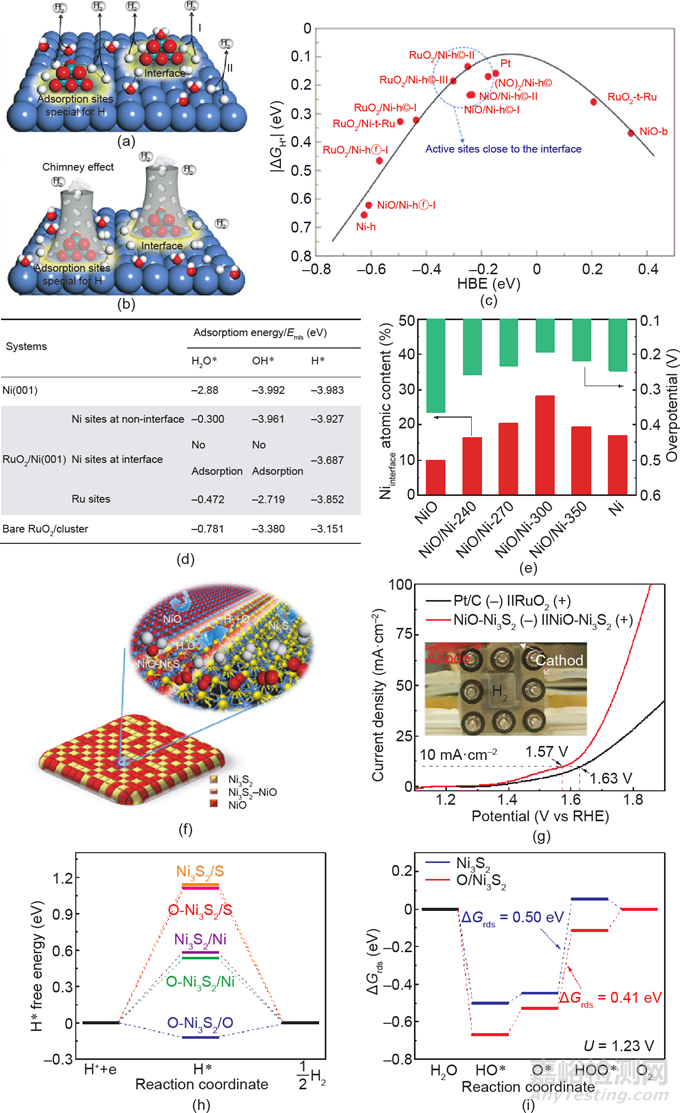
圖19 (a)RuO2/Ni復(fù)合催化劑中兩種可能的HER路徑圖示���;(b)在金屬/金屬氧化物界面處的“煙囪效應(yīng)”���;(c)ΔGH*對(duì)氫吸附自由能的曲線���;(d)不同團(tuán)簇上H2O*、OH*和H*物種的吸附能���;(e)NiO/Ni樣品HER活性隨Niinterface含量的改變趨勢(shì);(f)單金屬NiO-Ni3S2雜化納米片的水裂解圖示���;(g)NiO-Ni3S2及Pt/C和RuO2電極組合的水裂解極化曲線;Ni3S2和O-Ni3S2表面的氫吸附自由能(h)和OER各步的反應(yīng)自由能圖(i)
空位形成是界面材料的催化性能提升的另一個(gè)重要因素���。Xi等報(bào)道了一個(gè)典型FeS2/CoS2雜化納米片(FeS2/CoS2-NSs)用于催化水電解。在合成過(guò)程中���,通過(guò)采用共沉淀法制備了CoFe2O4納米粒子���,后通過(guò)硫化將其轉(zhuǎn)化為FeS2和CoS2相,并產(chǎn)生包含缺陷位點(diǎn)的界面���。電子順磁共振(EPR)譜顯示���,F(xiàn)eS2/CoS2-NSs復(fù)合材料在g=2.007處具有較強(qiáng)的EPR信號(hào),這表明其具有豐富的S空位���。擴(kuò)展X射線吸收精細(xì)結(jié)構(gòu)(EXAFS)進(jìn)一步研究樣品的局部結(jié)構(gòu)���,檢測(cè)到FeS2/CoS2-NSs中Fe的K邊EXAFS的峰強(qiáng)度明顯降低���,這說(shuō)明了Fe的配位缺陷���。Qu等在具有嵌入式結(jié)構(gòu)CeO2/NiO(Ce-NiO-E)或表面負(fù)載結(jié)構(gòu)CeO2/NiO(Ce-NiO-L)發(fā)現(xiàn)了類似的現(xiàn)象。NiO(Ni3+:62%���,氧缺陷:24%)���、Ce-NiO-L(Ni3+:69%,氧缺陷:26%)和Ce-NiO-E(Ni3+:71%���,氧缺陷:32%)中的Ni3+和氧缺陷的增加趨勢(shì)與活性趨勢(shì)密切相關(guān)���,表明界面區(qū)的空位對(duì)活性的增強(qiáng)有很大貢獻(xiàn)。事實(shí)上���,界面材料的電子結(jié)構(gòu)調(diào)控和空位并不獨(dú)立存在���,界面材料的催化性能可能同時(shí)受到這兩個(gè)因素的影響���。
(八)合金化
合金化是兩種或兩種以上金屬的金屬原子相互擴(kuò)散滲透���,或通過(guò)熔化、燒結(jié)或氣相沉積過(guò)程將非金屬元素添加到金屬中���。合金化是提高金屬催化劑性能的有效策略���,它不僅可以細(xì)化晶粒尺寸���,提高機(jī)械強(qiáng)度以及催化劑的比表面積���,還可選擇性地減少單一組分的用量,以降低催化劑的成本���。此外���,由于組分之間的協(xié)同作用���,通過(guò)加入其他元素形成合金���,可以改變金屬的催化活性和選擇性。根據(jù)Brewer-Engel價(jià)鍵理論���,將具有未填充d軌道的金屬和具有內(nèi)部成對(duì)d電子的金屬合金化可以調(diào)節(jié)合金表面的氫吸附能���,從而提高析氫活性���。Raj等采用電沉積技術(shù)制備了一系列鎳基二元復(fù)合材料���,在堿性溶液中的HER催化活性變化趨勢(shì)是:Ni-Mo>Ni-Zn>NiCo>Ni-W>Ni-Fe>Ni-Cr���。Ni-Mo合金因其優(yōu)異的催化活性被認(rèn)為是最有前途的HER催化材料。Zhang等利用磁控濺射技術(shù)在鎳基表面成功地構(gòu)建了一層尺寸均勻���、元素分布均勻的Ni-Mo合金納米棒,并發(fā)現(xiàn)其交換電流密度幾乎是單金屬催化劑的10倍���。研究表明���,Ni-Mo合金電極優(yōu)異的催化活性主要來(lái)自兩個(gè)方面:①在生長(zhǎng)過(guò)程中,雙組分金屬的晶粒細(xì)化導(dǎo)致比表面積增加���;②元素Ni和Mo的電負(fù)性差異導(dǎo)致電子在Mo周圍聚集���,從而形成電催化的協(xié)同作用���。
為了減少貴金屬的使用���,許多非貴金屬(Co���、Ni���、Fe���、Cu、V���、Cr、Mn���、Zn等)被用來(lái)與貴金屬結(jié)合形成合金作為電催化劑���。對(duì)不同PtM合金的ORR性能研究表明,其活性順序?yàn)椋篜tFe/C>PtCo/C>PtV/C>PtNi/C>Pt/C���,穩(wěn)定性趨勢(shì)為:Pt3Ir (111) >Pt3Co (111) >Pt3Ni (111) >Pt3Fe (111)���。此外,Stamenkovic等根據(jù)鉑合金的ORR活性與3d金屬的d帶中心位置���,發(fā)現(xiàn)了典型的火山關(guān)系(圖20)���。研究表明,Pt3M催化劑的ORR機(jī)制是O2解離或質(zhì)子/電子轉(zhuǎn)移到O2分子上���,最佳ORR催化劑的氧結(jié)合能應(yīng)比Pt弱���。Bampos等在酸性溶液中進(jìn)一步合成了一系列碳負(fù)載Pd-M(其中M=Ag、Co���、Cu���、Fe���、Ni或Zn)雙金屬催化劑���,其活性變化趨勢(shì)為:PdZn/C>PdNi/C>Pt/C>PdAg/CPdCo/C>PdFe/C>PdCu/C>Pd/C。其中���,最優(yōu)的PdZn/C在0.35~0.5 V(vs. Ag/AgCl)電壓下的 ORR比活性比Pt/C高3倍���。除了金屬元素外,引入非金屬元素同樣可以提高合金的催化性能���。Sampath等制備了少層MoS2(1–x)Se2x合金���,其活性高于原始MoS2MoSe2���。通過(guò)調(diào)節(jié)MoS2(1–x)Se2x中Se/S的摻入比例���,系統(tǒng)地研究了催化劑的構(gòu)效關(guān)系,并發(fā)現(xiàn)MoS1.0Se1.0S顯示了最高的HER活性���。Gong和Xu課題組通過(guò)對(duì)Mo-S-Se合金的研究,發(fā)現(xiàn)了類似結(jié)果���。He等成功地控制了碳纖維上三元WS2(1–x)Se2x納米管中硫和硒的組成���,所造成的無(wú)序原子排列使WS2(1–x)Se2x具有優(yōu)異的電催化性能���。Jin等進(jìn)一步獲得了三元黃鐵礦型CoPS用于光/電化學(xué)析氫反應(yīng)。由于CoPS中P2–配體具有更高的給電子性質(zhì)���,該三元黃鐵礦CoPS具有更適中的氫吸附能力,使得其活性高于CoS2���。
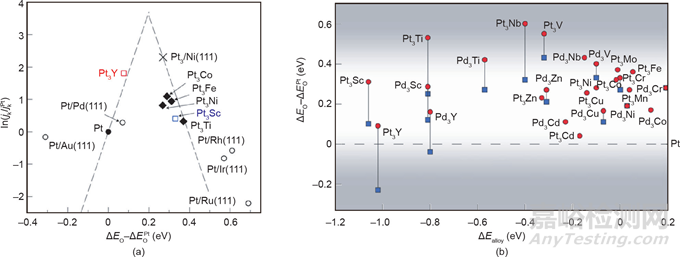
圖20 (a)測(cè)試所得動(dòng)力學(xué)電流密度和計(jì)算所得氧吸附能ΔEO之間的關(guān)系���;(b)鉑基合金表面鉑的ΔEO對(duì)合金能量圖
四、結(jié)論與展望
電化學(xué)氫-水轉(zhuǎn)化過(guò)程中的大部分能量耗散是由能源系統(tǒng)中的驅(qū)動(dòng)電化學(xué)反應(yīng)的活化能引起的���。由于催化材料對(duì)反應(yīng)活化能的重要影響,催化材料是高效能源系統(tǒng)的核心���。為了實(shí)現(xiàn)一個(gè)高效���、可持續(xù)的能源系統(tǒng),迫切需要加快開(kāi)發(fā)低成本���、高活性的電催化劑���。
一般來(lái)說(shuō),催化劑的性能取決于兩個(gè)主要因素:①給定區(qū)域內(nèi)活性位點(diǎn)的數(shù)目���;②每個(gè)活性位點(diǎn)的本征活性���。調(diào)整幾何結(jié)構(gòu)可有效地增加活性位點(diǎn)的密度。當(dāng)催化劑尺寸減小到納米尺度時(shí)���,催化劑具有高的比表面積���,并且增加催化活性晶面的暴露���。缺陷工程和非晶化也可以通過(guò)暴露熱力學(xué)不穩(wěn)定活性位或不飽和原子位點(diǎn)來(lái)增加活性位點(diǎn)的數(shù)量。提高活性位點(diǎn)的本征活性是提高活性的一個(gè)更基本但更困難的策略���,因?yàn)檫@涉及對(duì)催化材料電子結(jié)構(gòu)的精確調(diào)節(jié)���。本征活性的優(yōu)化需要對(duì)反應(yīng)的特性有基本理解和對(duì)具有目標(biāo)功能的催化劑的設(shè)計(jì)有深刻的見(jiàn)解。
文章所討論的典型實(shí)例(其催化性能總結(jié)見(jiàn)表2至表4)應(yīng)用理論與實(shí)驗(yàn)研究相結(jié)合���,通過(guò)元素?fù)诫s���、界面工程、晶相工程���、合金化等手段���,成功地調(diào)節(jié)了催化劑的本征活性。
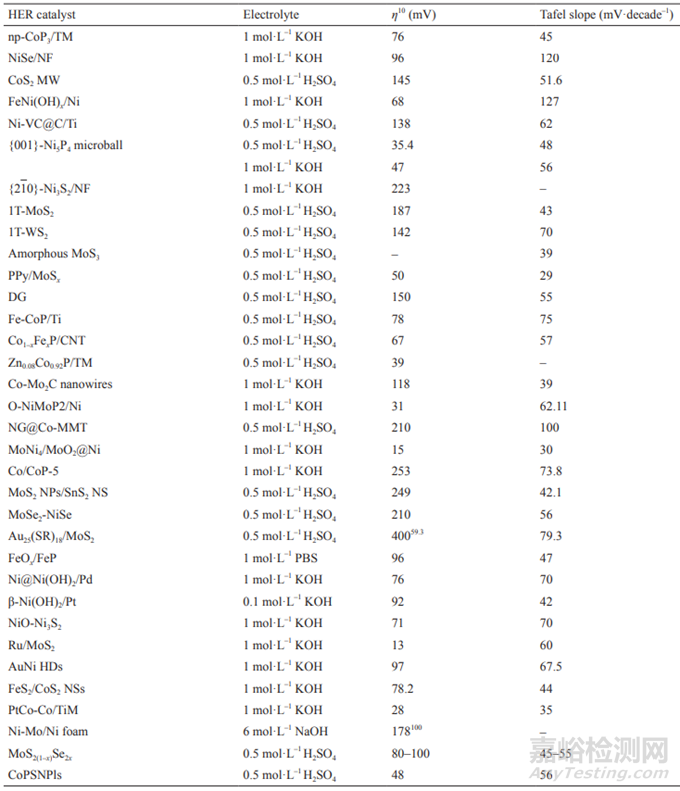
表2 典型HER催化劑性能
表3 典型OER催化劑性能
表4 典型非貴金屬ORR催化劑性能
表5 典型貴金屬ORR催化劑性能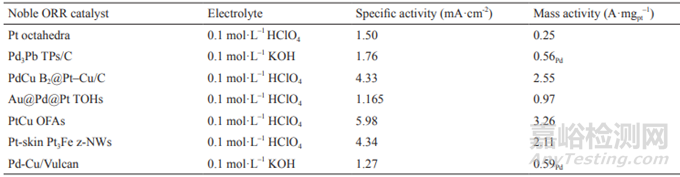
目前���,隨著化學(xué)合成技術(shù)的飛速發(fā)展,許多催化材料以其優(yōu)異的電催化性能被報(bào)道���。同時(shí)���,物理表征方法和理論計(jì)算的發(fā)展也為研究人員提供了更多的依據(jù)和指導(dǎo)���,使我們更好地了解催化劑的性能提升的機(jī)制���。然而���,現(xiàn)在電極催化劑的生產(chǎn)仍主要是通過(guò)傳統(tǒng)的試錯(cuò)工藝,而不是通過(guò)合理的設(shè)計(jì)���。這種研究模式不僅浪費(fèi)了社會(huì)資源和科研人員的精力���,而且極大地延緩了科學(xué)發(fā)展。因此���,迫切需要從最基本的層面發(fā)展一種面向性能的電催化劑設(shè)計(jì)策略,并使之能應(yīng)用于實(shí)際的催化劑設(shè)計(jì)合成中���。
這個(gè)目標(biāo)需要計(jì)算化學(xué)���、電催化化學(xué)和合成化學(xué)協(xié)同工作���;然而不幸的是���,目前這三個(gè)領(lǐng)域都需要改進(jìn)。目前的理論計(jì)算模型還不完善���,因?yàn)榇蠖鄾](méi)有考慮離子強(qiáng)度���、雙層效應(yīng)、溶劑化效應(yīng)等因素的影響���,難以準(zhǔn)確反映催化材料表面的實(shí)際反應(yīng)過(guò)程���。此外,盡管這些氫和氧參與反應(yīng)的機(jī)制已經(jīng)被廣泛研究���,但在不同催化劑表面上的實(shí)際反應(yīng)機(jī)制仍然是個(gè)謎���。事實(shí)上,由于大多數(shù)催化劑的表面結(jié)構(gòu)在電催化過(guò)程中一直在變化���,因此很難確定實(shí)際的活性中心���。先進(jìn)的理論計(jì)算和實(shí)驗(yàn)表征的結(jié)合將帶來(lái)新的進(jìn)展,這將有助于我們?cè)诜肿铀缴侠斫怆娀瘜W(xué)反應(yīng)機(jī)制和催化劑的動(dòng)態(tài)演化���。
最后���,面向功能的催化劑制備仍是一個(gè)重大挑戰(zhàn)。現(xiàn)有的材料合成技術(shù)能夠在納米尺度上對(duì)特定催化劑的理化性質(zhì)進(jìn)行一定程度的調(diào)控���,但利用可控合成技術(shù)在原子尺度上調(diào)控催化劑的電子結(jié)構(gòu)還不成熟���。發(fā)展適用性強(qiáng)、規(guī)?��;a(chǎn)的可控合成方法是未來(lái)研究的另一個(gè)熱點(diǎn)���。
改編原文:
Lishan Peng, Zidong Wei. Catalyst Engineering for Electrochemical Energy Conversion from Water to Water: Water Electrolysis and the Hydrogen Fuel Cell[J]. Engineering,2020,6(6):653-679.