時間僅剩3個月�����! 2024年5月26日前��,遺留器械應(yīng)當提交MDR符合性評估申請并建立質(zhì)量管理體系����,否則無法享受過渡期延長��。
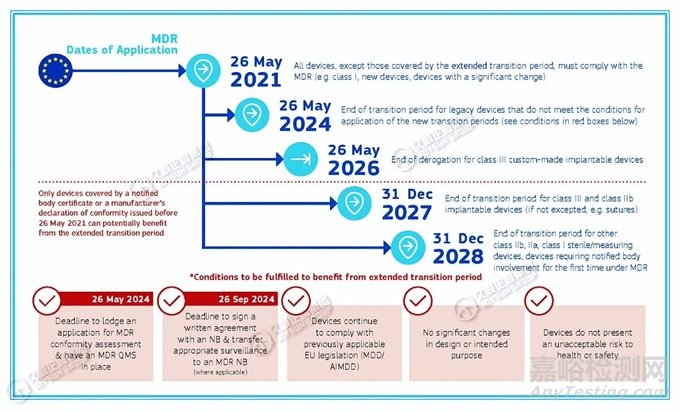
MDR法規(guī)過渡期延長的時間要求
歐盟《MDR過渡期時間表》提出:2024年5月26日����,不滿足新過渡期應(yīng)用條件的遺留器械的過渡期結(jié)束��;
從過渡期延長中受益所需滿足的條件有:
· 2024年5月26日:提交MDR符合性評估申請并建立MDR質(zhì)量管理體系的截止日期��;
· 2024年9月26日:與公告機構(gòu)簽署書面協(xié)議并將適當?shù)谋O(jiān)督轉(zhuǎn)移至MDR公告機構(gòu)(如適用)的截止日期��;
· 器械繼續(xù)遵守先前適用的歐盟法規(guī)(MDD/ AIMDD);
· 設(shè)計或預(yù)期目的無重大變化�����;
· 器械不會對健康或安全構(gòu)成不可接受的風險����。
遺留器械的定義
· 在2021年5月26日前,MDD指令下的I類醫(yī)療器械已擬訂符合性聲明�����,MDR法規(guī)下其符合性評估程序需要公告機構(gòu)參與的����;
· 在2021年5月26日前,按照MDD指令或AIMDD指令已取得CE標志����。
MDR法規(guī)對質(zhì)量管理體系的要求
按照MDR要求,制造商需要基于MDR要求建立����、運行并保持質(zhì)量管理體系。
2024年5月26日前����,所有遺留器械的質(zhì)量體系建設(shè)必須符合MDR下EN ISO 13485:2016��。
針對遺留器械��,MDR法規(guī)新增許多質(zhì)量體系要求:包括上市后監(jiān)督�����、市場監(jiān)督�����、警戒系統(tǒng)��、經(jīng)濟運營商和器械登記等����,對此制造商最晚不遲于2024年5月26日已建立質(zhì)量管理體系����。