美國(guó) FDA 對(duì)于藥品前沿技術(shù)的發(fā)展進(jìn)行快速響應(yīng)����,致力于促進(jìn)創(chuàng)新產(chǎn)品的快速上市����,最大限 度的滿足公眾健康需求����。 本文擬通過對(duì)美國(guó) FDA 藥品前沿技術(shù)監(jiān)管領(lǐng)域的相關(guān)政策、行動(dòng)計(jì)劃����、技術(shù)指南 以及實(shí)施程序等進(jìn)行分析總結(jié),以期為我國(guó)前沿技術(shù)領(lǐng)域醫(yī)藥產(chǎn)品的科學(xué)監(jiān)管提供借鑒�����。
科學(xué)和技術(shù)的進(jìn)步為開發(fā)創(chuàng)新醫(yī)療產(chǎn)品提供了非凡的機(jī)遇����,這些產(chǎn)品的問世為患者帶來更好的治 療體驗(yàn)、提高患者生存質(zhì)量甚至挽救更多生命��。 為了應(yīng)對(duì)技術(shù)進(jìn)步的速度快于新產(chǎn)品風(fēng)險(xiǎn)?獲益科學(xué) 評(píng)價(jià)的發(fā)展速度�����,鼓勵(lì)藥品前沿技術(shù)的發(fā)展與應(yīng)用��, 美國(guó)FDA 時(shí)刻保持先進(jìn)性和國(guó)際引領(lǐng)地位��,開發(fā)了 一系列創(chuàng)新的監(jiān)管新工具�����、新方法和新標(biāo)準(zhǔn)����,將更多 的藥品前沿技術(shù)推向市場(chǎng),確保已上市藥品的安全 有效�����,持續(xù)保障和促進(jìn)公眾健康����。 本文主要介紹美國(guó) FDA 在先進(jìn)制造(advanced manufacturing)、藥物開發(fā)工具�����、再生醫(yī)學(xué)先進(jìn)療法 3 個(gè)方面的監(jiān)管實(shí)踐經(jīng)驗(yàn)�����,以期為我國(guó)相關(guān)前沿技術(shù)領(lǐng)域的監(jiān)管提供參考。
1�����、 先進(jìn)制造
先進(jìn)制造已成為全球趨勢(shì)��,引起許多國(guó)外監(jiān)管 機(jī)構(gòu)的高度重視��。 美國(guó) FDA 投入大量資金用于先 進(jìn)制造相關(guān)項(xiàng)目�����,持續(xù)與業(yè)界合作�����,促進(jìn)先進(jìn)制造技 術(shù)的實(shí)施����,讓更多患者獲益。
1.1 先進(jìn)制造的含義 先進(jìn)制造是提高藥品質(zhì)量����、解決藥品短缺問題并加快藥品上市時(shí)間的新醫(yī)療產(chǎn)品制造技術(shù)的統(tǒng) 稱,這些技術(shù)整合新的技術(shù)方法,以創(chuàng)新的方式使用 現(xiàn)有技術(shù)��,或者在沒有明確的最佳實(shí)踐或經(jīng)驗(yàn)的新 領(lǐng)域應(yīng)用生產(chǎn)方法����。 先進(jìn)制造技術(shù)包括:1 連續(xù)制 造( continuous manufacturing�����,CM) ��。 CM 提供了一種更快�����、更可靠的制藥方式��。 美國(guó) FDA 正在促進(jìn)這種 方法廣泛使用����。 CM將傳統(tǒng)的逐步制造流程集成到 基于現(xiàn)代流程監(jiān)控和控制的單一系統(tǒng)中。 在 CM 過 程中��,產(chǎn)品是隨著時(shí)間的推移而制造的,因此制藥企 業(yè)可以輕松控制生產(chǎn)的產(chǎn)品數(shù)量以滿足需求。 這些 高效����、集成的連續(xù)系統(tǒng)僅需要更小的占地面積即可 運(yùn)行����。 2 增材制造(additive manufacturing,常被簡(jiǎn) 稱 3D 打印��,即 3DP):能夠通過重復(fù)分層材料將數(shù) 字 3D 模型制作成實(shí)體零件�����。 這些方法可用于所有 醫(yī)療產(chǎn)品����。 3DP 可以創(chuàng)建特定患者的醫(yī)療設(shè)備,如植入物�����、手術(shù)指南或解剖學(xué)模型�����。 同樣�����,3DP 的固體 藥物產(chǎn)品可以制成各種形狀�����、規(guī)格以及活性和非活 性成分的分布����。 同時(shí)正在研究組織工程的 3DP 構(gòu) 造物可以推進(jìn)再生醫(yī)學(xué)��。 這種方法提供可以生產(chǎn)出 適合患者個(gè)人需求的醫(yī)療產(chǎn)品的獨(dú)特機(jī)會(huì)�����。 3DP 工 藝的能力和便攜性也使復(fù)雜和先進(jìn)的醫(yī)療產(chǎn)品在偏 遠(yuǎn)或艱苦條件下的分布式制造成為可能。
1.2 先進(jìn)制造計(jì)劃
根據(jù)美國(guó) FDA 藥物評(píng)價(jià)與研究中心(CDER)藥品質(zhì)量辦公室(OPQ)2021 年度報(bào)告[1] ����,OPQ 正 在努力通過 3 個(gè)關(guān)鍵舉措實(shí)現(xiàn)建立先進(jìn)制造技術(shù)。 1.2. 1 新 興 技 術(shù) 計(jì) 劃 ( emerging technology program��, ETP) 為了促進(jìn)制造生產(chǎn)中新技術(shù)的使用����,CDER 于2013年啟動(dòng)新興技術(shù)團(tuán)隊(duì)(ETT)的建立[2]。 2014 年底����,CDER 制定了 ETP ,制藥企業(yè)代表可以在 企業(yè)提交藥品上市申請(qǐng)之前與美國(guó) FDA 互動(dòng)����,以討 論新型先進(jìn)制造技術(shù)開發(fā)過程中的技術(shù)和監(jiān)管問 題。 盡管該計(jì)劃最初側(cè)重于防止對(duì)新制造技術(shù)的審 評(píng)延遲����,但制藥商無需有已提交的注冊(cè)申請(qǐng)即可參 與該計(jì)劃。 ETP 不僅涵蓋 CM��,還涵蓋無菌技術(shù)、模 塊化制造�����、建模����、人工智能 / 機(jī)器學(xué)習(xí)和智能制造等 其他領(lǐng)域。 為了滿足業(yè)界申請(qǐng)的需求�����,進(jìn)一步加速 新制藥技術(shù)的采納�����,CDER 正在為其新興技術(shù)項(xiàng)目 增加人員��,并逐步將新技術(shù)探索“ 畢業(yè)” 推進(jìn)到標(biāo)準(zhǔn) 質(zhì)量評(píng)價(jià)路徑中����。 2021 年����,為進(jìn)一步加強(qiáng)現(xiàn)有的流 程和結(jié)構(gòu)����,CDER 將 ETP 升級(jí)至 ETP 2.0�����,該計(jì)劃包 括 13 個(gè)優(yōu)先領(lǐng)域:畢業(yè)�����、知識(shí)管理和轉(zhuǎn)移��、管治��、使 用��、參與����、溝通、技術(shù)和工具�����、技能和培訓(xùn)�����、工作量、管 理����、策略以及意識(shí)[3] 。
在 ETP 中��,畢業(yè)部分將分 3 個(gè)步驟進(jìn)行:第 1 步����,行業(yè)在準(zhǔn)備監(jiān)管申報(bào)時(shí)申請(qǐng)對(duì)新興技術(shù)的意見 和反饋;第 2 步,新興技術(shù)團(tuán)隊(duì)隨后將與行業(yè)合作����, 討論、確定和解決與新技術(shù)開發(fā)和實(shí)施相關(guān)的技術(shù)和監(jiān)管問題;第 3 步����,該技術(shù)將不再被視為“ 新興” 技術(shù)��,而是通過標(biāo)準(zhǔn)的質(zhì)量評(píng)價(jià)路徑審評(píng)����,稱之為畢 業(yè)����。 畢業(yè)提供了 3 個(gè)好處:1 使美國(guó) FDA 能夠獲 得足夠的技術(shù)經(jīng)驗(yàn)�����,并相信行業(yè)將提交成功的未來 申請(qǐng)�����。 2 有能力接受未來的新興技術(shù)��,跟上行業(yè)創(chuàng) 新的步伐��。 3 能夠在繼續(xù)滿足《 處方藥使用者付費(fèi) 法案》(PDUFA)目標(biāo)日期的同時(shí)審評(píng)更多申請(qǐng)��。 目 前 ETP 2.0 已分階段實(shí)施����。 美國(guó) FDA 的數(shù)據(jù)顯示, 雖然 CM 應(yīng)用仍然是 ETP 最常見的申請(qǐng)類型��,但該 計(jì)劃越來越多地應(yīng)用于新型無菌技術(shù)����。
1.2.2 先進(jìn)制造評(píng)估監(jiān)管框架計(jì)劃(framework for reg? ulatory advanced manufacturing evaluation�����,F(xiàn)RAME) CDER 制定了 FRAME 計(jì)劃�����,以支持為患者帶來獲益 的先進(jìn)制造技術(shù)[4] �����。 FRAME 計(jì)劃指的是 OPQ 資助 美國(guó)國(guó)家科學(xué)��、工程和醫(yī)學(xué)研究院(NASEM)�����,幫助 收集制藥企業(yè)對(duì)于未來 5 ~ 10 年先進(jìn)制造技術(shù)的投 入��,通過一系列研討會(huì)��,匯集了行業(yè)、學(xué)術(shù)界和政府 專家對(duì)于藥品制造的未來發(fā)展共識(shí)����,形成相關(guān)報(bào)告, 明確了美國(guó) FDA 未來可能接收的新技術(shù)申報(bào)�����,這些 技術(shù)包括:端到端連續(xù)制造�����,一種整合原料藥和制劑 的過程;便攜式和模塊化分布式制造平臺(tái)��,即所謂的 按需藥房(pharmacy on demand)[5] ;制造中的人工 智能或高級(jí)建模方法�����。 2021 年�����,生物制品評(píng)價(jià)和研 究中心( Center for Biologics Evaluation and Research����, CBER)和 CDER 聯(lián)合成立了內(nèi)部藥品和生物制藥 促進(jìn)中心,以進(jìn)一步調(diào)整美國(guó) FDA 與 FRAME 相關(guān) 的思維和政策��,促進(jìn)對(duì)使用先進(jìn)制造技術(shù)的藥品申 請(qǐng)的審評(píng)[6] 。 作為計(jì)劃的一部分��,CDER 計(jì)劃通過 討論文件和研討會(huì)征求利益相關(guān)者的意見��,制訂或 修訂相關(guān)指南�����,并計(jì)劃協(xié)調(diào)國(guó)際指南�����。
1.2.3 質(zhì)量管理成熟度( quality management maturity��,QMM)評(píng)級(jí) 2022 年 4 月 5 日��,OPQ 發(fā)布了 《QMM 白皮書》[7] ����,將 QMM 描述為“藥品制造商擁 有一致、可靠和穩(wěn)健的業(yè)務(wù)流程以實(shí)現(xiàn)質(zhì)量目標(biāo)并 促進(jìn)持續(xù)改進(jìn)所達(dá)到的狀態(tài)”�����。 白皮書指出產(chǎn)品質(zhì) 量高��、供應(yīng)鏈彈性低、藥品持續(xù)短缺的現(xiàn)狀��,提倡美 國(guó) FDA 與利益相關(guān)者建立 QMM 計(jì)劃�����,建立科學(xué)基 金����,鼓勵(lì)利益相關(guān)者參與��。 根據(jù)美國(guó)多機(jī)構(gòu)聯(lián)邦特 別工作組報(bào)告顯示����,許多藥品短缺的根本原因是缺 乏激勵(lì)措施,促進(jìn)制造商在滿足現(xiàn)行良好生產(chǎn)規(guī)范 (cGMP)基礎(chǔ)上����,具備努力開發(fā)成熟的質(zhì)量管理體 系的動(dòng)力。 因此��,CDER 提出 QMM 評(píng)級(jí)系統(tǒng)��,幫助 激勵(lì)藥品制造商在其設(shè)施中實(shí)現(xiàn)質(zhì)量管理��。 在缺乏QMM 評(píng)級(jí)透明度的情況下,價(jià)格競(jìng)爭(zhēng)和成本最小化 是市場(chǎng)驅(qū)動(dòng)的關(guān)鍵因素�����,特別是對(duì)于仿制藥�����,而積極 投資以避免未來藥物短缺的制藥商卻沒有直接回 報(bào)��。 成熟的質(zhì)量管理是以績(jī)效和以患者為中心的體 系確定需要改進(jìn)的領(lǐng)域并實(shí)施有效的變革�����。 QMM 評(píng)級(jí)系統(tǒng)可以向監(jiān)管機(jī)構(gòu)和采購商通報(bào)藥品生產(chǎn)設(shè) 施的性能和穩(wěn)健性����,并增加患者對(duì)藥品可獲得性的 信心。 QMM 評(píng)級(jí)還可以提高制造商在批準(zhǔn)后進(jìn)行 生產(chǎn)變更的靈活性�����,減少監(jiān)管監(jiān)督����,激勵(lì)持續(xù)改進(jìn)����。
美國(guó) FDA 表示����,要使這樣的評(píng)級(jí)系統(tǒng)發(fā)揮作用�����,市 場(chǎng)需要獎(jiǎng)勵(lì)制藥設(shè)施 QMM 評(píng)分較高的產(chǎn)品��,在采 購決策中使用 QMM 評(píng)級(jí)應(yīng)該激勵(lì)長(zhǎng)期的持續(xù)改 進(jìn)��,但不會(huì)在短期內(nèi)將制藥商擠出市場(chǎng)或顯著提高采購成本��。
1.3 美國(guó) FDA 發(fā)布 / 實(shí)施的相關(guān)技術(shù)指南
美國(guó) FDA 與制藥企業(yè)合作制定了一系列先進(jìn) 制造技術(shù)相關(guān)技術(shù)指南�����,闡明支持使用先進(jìn)制造技術(shù)的產(chǎn)品所需的法規(guī)和數(shù)據(jù)要求����,見表 1。
2��、藥物開發(fā)工具
DDT的含義:DDT 是指促進(jìn)藥物開發(fā)的方法、材料或措施��,用于降低開發(fā)美國(guó) FDA 監(jiān)管產(chǎn)品的時(shí)間��、復(fù)雜性或 成本�����,同時(shí)提高產(chǎn)品開發(fā)的可靠性和穩(wěn)健性�����。 DDT 包括新型生物標(biāo)志物的開發(fā)�����、通過推進(jìn)藥效學(xué)( PD) 生物標(biāo)志物的使用來提高生物類似物開發(fā)和批準(zhǔn)的 效率����、新技術(shù)用于提高非臨床研究的可預(yù)測(cè)性,并替 代(replacement)�����、減少(reduction)和改進(jìn)(refine? ment)動(dòng)物實(shí)驗(yàn)(以下簡(jiǎn)稱 3R)����。 目前美國(guó) FDA 的 藥物開發(fā)工具認(rèn)證計(jì)劃有 CDER 生物標(biāo)志物資格�����、 臨床結(jié)果評(píng)估資格����、新藥創(chuàng)新科學(xué)和技術(shù)方法 ( ISTAND) 試點(diǎn)計(jì)劃�����、設(shè)備和輻射衛(wèi)生中心( CDRH) 非臨床評(píng)估模型等�����。
2.2 美國(guó) FDA 藥物開發(fā)工具的監(jiān)管發(fā)展歷程
2.2.1 關(guān)鍵路徑計(jì)劃( critical path initiative�����,CPI) 2004 年��,美國(guó) FDA 提出 CPI��,其中指出生物醫(yī)學(xué)上 諸多技術(shù)進(jìn)步尚未顯著助力藥物和新療法的研發(fā)����, CPI 的一個(gè)重要作用是將先進(jìn)的生物醫(yī)學(xué)技術(shù)作為 新工具運(yùn)用在藥物開發(fā)過程中,加速安全��、有效的新 醫(yī)藥產(chǎn)品研發(fā)上市�����。
2.2.2 生物標(biāo)志物資格認(rèn)定計(jì)劃( biomarkers quali? fication program����,BQP) 2009 年美國(guó) FDA 建立了 BQP,為行業(yè)建立生物標(biāo)志物研發(fā)和認(rèn)定平臺(tái)����,給予 生物標(biāo)志物資格認(rèn)定并公開提供支持信息,促進(jìn)獲 得資格認(rèn)定的生物標(biāo)志物資格的使用�����,鼓勵(lì)藥物研 發(fā)和監(jiān)管決策中新的生物標(biāo)志物的認(rèn)定����。 一旦生物 標(biāo)志物獲得資格認(rèn)定,發(fā)起人可將其用于符合特定應(yīng)用場(chǎng)景的藥物研發(fā)計(jì)劃,而無須 CDER 重新審查 相關(guān)支持性信息�����。
2.2.3 藥物開發(fā)工具資格認(rèn)定程序(qualification process for drug development tools����, QPDDT ) 2010 年,美國(guó) FDA 發(fā)布《 面向行業(yè)和 FDA 的藥物開發(fā)工 具資格認(rèn)定程序指南草案》��,并于 2014 年正式發(fā) 布[8] �����。 該指南由 CDER 生物標(biāo)志物資格認(rèn)定工作 組制定��。
2015 年��,CDER 建立了 QPDDT��, 通過該程序鼓 勵(lì)申辦者公開獲得認(rèn)定的創(chuàng)新藥物研發(fā)工具��,促進(jìn) 生物標(biāo)志物臨床結(jié)局評(píng)價(jià)工具(biomarkers clinical outcome assessments)�����、動(dòng)物模型(animal models)等 創(chuàng)新性藥物開發(fā)工具在藥物研發(fā)中的推廣使用��,減 少新型開發(fā)工具的重復(fù)審評(píng)����。 生物標(biāo)志物日益成為 治療特定疾病的醫(yī)藥產(chǎn)品研發(fā)的重要組成部分,因 此 QPDDT 中最重要的就是生物標(biāo)志物資格認(rèn)定項(xiàng) 目��,醫(yī)藥產(chǎn)品研發(fā)中作為有效指標(biāo)的生物標(biāo)志物�����,經(jīng) 該程序獲美國(guó) FDA 審評(píng)通過后即認(rèn)定為合格的生 物標(biāo)志物����。
2016 年《21 世紀(jì)治愈法案》 促進(jìn)國(guó)家精準(zhǔn)醫(yī)學(xué) 行動(dòng)的倡議的落實(shí),在對(duì)疾病的分子水平理解的基 礎(chǔ)上����,進(jìn)一步推動(dòng)疾病預(yù)防、診斷和治療產(chǎn)品的研 發(fā)��。 《聯(lián)邦食品��、藥品和化妝品法案》(FD&C)新增 第507節(jié)“藥物開發(fā)工具資格”認(rèn)定條款�����。 2019年 12 月《 藥物開發(fā)工具的資格認(rèn)定程序指南草案》 全 面修訂后再次同名發(fā)布。
2.2.4 ISTAND 試點(diǎn)計(jì)劃 為了支持����、簡(jiǎn)化可能加 速藥品開發(fā)的新技術(shù),美國(guó) FDA 于 2020 年 11 月 30日啟動(dòng)了 ISTAND 試點(diǎn)計(jì)劃��,并于 2021 年繼續(xù)實(shí)施 該計(jì)劃����。 該試點(diǎn)為尚無監(jiān)管途徑的新穎方法提供提 交途徑,旨在鼓勵(lì)開發(fā)現(xiàn)有 DDT 資格認(rèn)證計(jì)劃范圍 外但對(duì)藥物開發(fā)有益的 DDT�����,支持開發(fā)可行的新型 藥物開發(fā)方法��。 ISTAND 由藥品審評(píng)科學(xué)辦公室 (Office of Drug Evaluation Sciences��,ODES) 領(lǐng)導(dǎo)�����,鼓 勵(lì)開發(fā)和使用新的 DDTs�����,這些 DDTs 的特點(diǎn)是整合 了科學(xué)和技術(shù)的最新進(jìn)展����,但目前沒有明確路徑以 及 DDT 確認(rèn)程序。 申辦者通過 ISTAND 試點(diǎn)項(xiàng)目 的提交程序提交相關(guān)申請(qǐng)�����,一旦合格��,DDT 就可以 在合格的使用范圍內(nèi)用于相關(guān)藥物的開發(fā)����。 此外,合格的 DDT 一般可被納入新藥 IND����,NDA 或 BLA 中,而不需要美國(guó) FDA 重新考慮和確認(rèn)其適用性����。
同時(shí),通過 ISTAND 試點(diǎn)計(jì)劃����,對(duì)于不適合現(xiàn)有 的 DDT 資格認(rèn)證模式但仍對(duì)藥物開發(fā)具有潛在價(jià) 值的其他項(xiàng)目��,美國(guó) FDA 監(jiān)管人員可以與申辦者召 開一系列會(huì)議��,提出相關(guān)研究意見;舉行公開會(huì)議��, 征求對(duì)新方法的意見;編寫“白皮書”����,提出實(shí)施新 型 DDT 的考慮;制定指南����,說明美國(guó) FDA 在藥物開 發(fā)中使用該新工具的立場(chǎng);或以其他方式向美國(guó) FDA 提供意見和建議,以支持 DDT 的進(jìn)一步發(fā)展�����。 2.2.5 新替代方法計(jì)劃 2011 年����,美國(guó)國(guó)立衛(wèi)生研 究院( NIH) 、美國(guó) FDA 與美國(guó)國(guó)防高級(jí)研究計(jì)劃局 (DARPA)聯(lián)合項(xiàng)目立項(xiàng)��,推出了“微生理系統(tǒng)”計(jì) 劃(MPS 計(jì)劃)�����,把器官芯片(organs?on?chips��,OoC) 技術(shù)的開發(fā)和應(yīng)用上升到國(guó)家戰(zhàn)略層面����。 2022 年 6 月,通過《食品和藥品修正案》����,將器官芯片和微生理系統(tǒng)作為獨(dú)立的藥物非臨床試驗(yàn)評(píng)估體系納入法 案。 2022 年 6 月 14 日����,美國(guó) FDA 發(fā)布的《促進(jìn)監(jiān)管 使用的替代方法》 報(bào)告,闡述了美國(guó) FDA 提議的新 替代方法計(jì)劃�����,包括美國(guó) FDA 提議的新替代方法計(jì) 劃����、各產(chǎn)品領(lǐng)域的具體考慮、新的替代方法應(yīng)用研究 和在監(jiān)管受理案例(心臟安全��、發(fā)育和生殖毒性)、 摘要和后續(xù)計(jì)劃��。 2022 年 12 月��,美國(guó)參眾兩院共 同通過美國(guó) FDA 現(xiàn)代化法案 2.0(FDA Moderniza? tion Act 2.0) ��,取消 FD&C 對(duì)新藥和仿制藥進(jìn)行動(dòng)物 實(shí)驗(yàn)的強(qiáng)制要求�����,旨在未來幾年里大幅減少動(dòng)物實(shí) 驗(yàn)的使用�����,推進(jìn)包括器官芯片在內(nèi)的多樣化臨床前 測(cè)試模型�����,減少動(dòng)物的使用;將非臨床實(shí)驗(yàn)定義為細(xì) 胞實(shí)驗(yàn)�����、器官芯片和微生理系統(tǒng)��、計(jì)算機(jī)模型����、其他 非動(dòng)物或人生物學(xué)方法和動(dòng)物實(shí)驗(yàn),提供了器官芯 片和微生理系統(tǒng)在內(nèi)的可作為藥物注冊(cè)支持性數(shù)據(jù) 的一種可能����。 2023 財(cái)年,總統(tǒng)預(yù)算提議為實(shí)施跨機(jī) 構(gòu)的新替代方法計(jì)劃提供資金����,以鼓勵(lì)采用新替代 方法進(jìn)行監(jiān)管,以替代�����、減少和改進(jìn)動(dòng)物實(shí)驗(yàn)并提高 非臨床試驗(yàn)的可預(yù)測(cè)性��,簡(jiǎn)化美國(guó) FDA 監(jiān)管產(chǎn)品 的開發(fā)��,更快����、更高效地將產(chǎn)品帶給美國(guó)公眾和患 者,確保產(chǎn)品安全��、有效��、可信賴。
2.3 美國(guó) FDA 發(fā)布 / 實(shí)施的相關(guān)技術(shù)指南
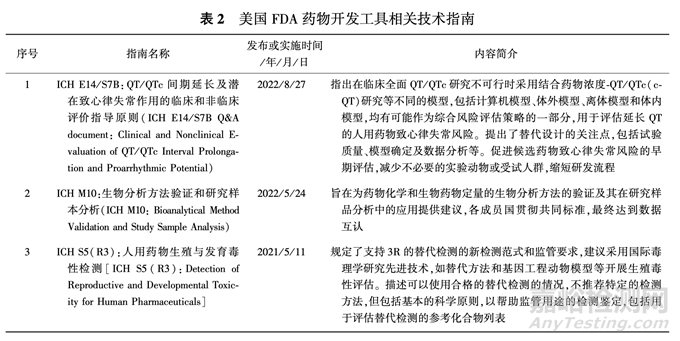
美國(guó) FDA 通過監(jiān)管項(xiàng)目的實(shí)施��,推動(dòng)制定一系 列技術(shù)指南�����,加速新的藥物開發(fā)工具的應(yīng)用與采納��, 滿足制藥技術(shù)發(fā)展需要��,為監(jiān)管決策提供參考��,包括 美國(guó) FDA 與利益相關(guān)者共同制定的技術(shù)指南以及監(jiān)管接受的IC H指南����,見表2.
3、 再生醫(yī)學(xué)先進(jìn)療法(regenerative medicine ad? vanced therapies�����,RMAT)
3.1 RMAT 含義
美國(guó)于 2016 年 12 月發(fā)布《21 世紀(jì)治愈法案》 中提出 RMAT����,為有生命危險(xiǎn)的患者提供接受尖端 醫(yī)療的創(chuàng)新審批途徑。 通過 RMAT 認(rèn)定的醫(yī)療產(chǎn) 品,可以在臨床試驗(yàn)早期更頻繁地與美國(guó) FDA 交 流��,獲得優(yōu)先審查和加速審批�����,能夠更快更精簡(jiǎn)地獲 批�����,從而縮短產(chǎn)品上市時(shí)間�����。 RMAT 認(rèn)定需要滿足 以下 3 個(gè)條件:1 該藥物是一種再生醫(yī)學(xué)療法�����,其定 義為細(xì)胞療法�����、治療性組織工程產(chǎn)品��、人體細(xì)胞和組 織產(chǎn)品����,或使用此類療法或產(chǎn)品的任何組合產(chǎn)品,《 公 共衛(wèi)生服務(wù)法》第 316 條(PHSA 361)和《聯(lián)邦法規(guī) 21 章》第 1271 條(21CFR Part 1271) 單獨(dú)規(guī)定的除外��。 2 該藥物在治療��、改變��、逆轉(zhuǎn)或治愈嚴(yán)重或危及生命 的疾病或狀況�����。 3 初步的臨床證據(jù)表明�����,該藥物有 潛力解決此類疾病或狀況未得到滿足的醫(yī)療需求��。
RMAT 資格認(rèn)定申請(qǐng)必須與 IND 或 IND 的修 正補(bǔ)充申請(qǐng)同時(shí)向 CBER 下屬的治療產(chǎn)品辦公室 (Office of Therapeutic Products�����,OTP) 提交�����,包括:該 藥物是否符合 RMAT 的定義;藥物如何達(dá)到預(yù)期治 療標(biāo)準(zhǔn);藥物有潛力解決此類疾病未滿足的醫(yī)療需求的初步臨床證據(jù)。
3.2 RMAT 產(chǎn)品的監(jiān)管進(jìn)展
3.2.1 RMAT 的特點(diǎn) RMAT 產(chǎn)品提供了全新的治 療途徑��,對(duì)亟待解決的醫(yī)療需求作出貢獻(xiàn)�����。 RMAT產(chǎn)品具備以下特征:1 原料細(xì)胞的質(zhì)量參差不齊��, 難以評(píng)估�����。 2 考慮到目標(biāo)疾病的特點(diǎn)����,可入組臨床 試驗(yàn)的患者數(shù)量有限�����。 3 如果涉及手術(shù)移植�����,在使 用對(duì)照組進(jìn)行比較實(shí)驗(yàn)時(shí)存在倫理問題�����。 4 可能 需要長(zhǎng)時(shí)間收集和評(píng)價(jià)數(shù)據(jù),以確認(rèn)有效性和安全 性����。 因此,按照目前的常規(guī)藥物開發(fā)規(guī)模�����,RMAT 產(chǎn) 品的臨床試驗(yàn)可行性顯著降低����,且需要很長(zhǎng)一段時(shí) 間才能將產(chǎn)品投入國(guó)際使用,研發(fā)和監(jiān)管的難度較 常規(guī)藥物更大����。
3.2.2 RMAT 的加速計(jì)劃 近年來,再生醫(yī)學(xué)領(lǐng)域 得到迅速發(fā)展��。 RAMT 具有治療嚴(yán)重疾病的潛力�����, 特別是針對(duì)醫(yī)療需求未得到滿足的患者����。 CBER 認(rèn) 識(shí)到再生醫(yī)學(xué)療法的重要性��,為了幫助確保 RMAT 產(chǎn)品獲得許可�����,2017 年 2 月發(fā)布了《 針對(duì)嚴(yán)重疾病 的再生醫(yī)學(xué)療法的加速審批程序》����,基于 RMAT 的 特點(diǎn)��,針對(duì)再生醫(yī)學(xué)療法提出 5 種加速計(jì)劃�����,包括 快速通道認(rèn)定�����、突破性療法認(rèn)定����、RMAT 認(rèn)定�����、優(yōu)先 審評(píng)認(rèn)定和加速批準(zhǔn)[9] 。 本文結(jié)合《 針對(duì)嚴(yán)重疾 病的藥物和生物制劑的加快審批程序》(2014 年 5 月)[10] ����,對(duì)比了 5 種加速程序在納入標(biāo)準(zhǔn)、政策獲 益�����、申請(qǐng)?zhí)峤粫r(shí)間以及美國(guó) FDA 回復(fù)時(shí)間等�����,見表 3�����。 其中��,RMAT 與突破性療法認(rèn)定之間的關(guān)鍵區(qū)別 是 RMAT 不要求申辦者提交研究用藥物與現(xiàn)有療 法相比的實(shí)質(zhì)性改善相關(guān)資料��。 如果某些產(chǎn)品獲得 RMAT 認(rèn)定并通過加速審批����,則可以優(yōu)先滿足某些 上市后要求����。
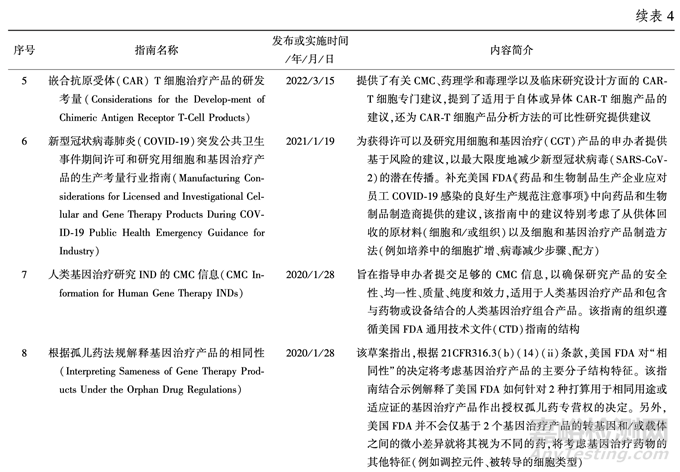
3.3 美國(guó) FDA 發(fā)布/ 實(shí)施的相關(guān)技術(shù)指南 為了促進(jìn) RMAT 產(chǎn)品的研發(fā)和上市進(jìn)程��,美 國(guó) FDA 基于現(xiàn)有的監(jiān)管認(rèn)知�����,發(fā)布了一系列技術(shù)指南�����,提出有效��、基于科學(xué)的指導(dǎo)原則��,以支持該 領(lǐng)域的研究�����。 截至目前�����,已發(fā)布了 17 項(xiàng)技術(shù)指南����, 見表 4。
3.4 美國(guó)RMAT實(shí)施現(xiàn)狀
RMAT 產(chǎn)品不僅要求具備創(chuàng)新性且能夠符合未滿足的臨床治療需求����。 根據(jù)美國(guó) FDA 官網(wǎng)數(shù) 據(jù),截至 2022 年 12 月 31 日��,美國(guó) FDA 共接收了 203 個(gè) RMAT 認(rèn)定產(chǎn)品的申請(qǐng)��,其中已同意 82 項(xiàng) (見表 5)��,覆蓋用于兒童先天性心臟病����、細(xì)胞淋巴 瘤等疾病治療領(lǐng)域[11] 。 由該項(xiàng)數(shù)據(jù)可見��,美國(guó) FDA 推行的一系列鼓勵(lì)政策和技術(shù)指南有效促進(jìn)了基因和細(xì)胞治療產(chǎn)品的申報(bào)����,但最終獲得 RMAT 資格認(rèn)定的產(chǎn)品僅 40%,認(rèn)定的門檻較高��。 美國(guó) FDA 目前僅批準(zhǔn)了 3 款 RMAT 認(rèn)定的產(chǎn)品�����,分別 是 Mallinckrodt 制藥公司的皮膚再生組織制品 Stratagraft(2017 年 7 月)、住友制藥母公司旗下的 Enzyvant 集團(tuán)公司的小兒先天性無胸腺癥治療藥 物 Rethymic 和百時(shí)美施貴寶的治療大 B 細(xì)胞瘤產(chǎn) 品 Breyanzi����。
4、 對(duì)我國(guó)的啟示
4.1 完善法律法規(guī)和指南框架 鼓勵(lì)和推進(jìn)新工具����、新方法、新標(biāo)準(zhǔn)的制定總結(jié)美國(guó)監(jiān)管實(shí)踐可以看出��,其支持前沿技術(shù) 的創(chuàng)新落實(shí)在國(guó)家戰(zhàn)略��、立法層面以及指南框架的 構(gòu)建等各方面:1 為了支持藥物研發(fā)工具的發(fā)展��, 美國(guó)將器官芯片和微生理系統(tǒng)作為獨(dú)立的藥物非臨 床試驗(yàn)評(píng)估體系納入《食品和藥品修正案》法案�����。 2 美國(guó)參眾兩院聯(lián)合通過美國(guó) FDA 現(xiàn)代化法案 2. 0����,取消 FD&C 對(duì)新藥和仿制藥進(jìn)行動(dòng)物實(shí)驗(yàn)的強(qiáng) 制要求,而我國(guó)目前尚無與《 食品和藥品修正案》 和 FD&C 類似的對(duì)于替代方法的相關(guān)要求��。 3 美國(guó) FDA 圍繞再生醫(yī)學(xué)療法制定了靈活的審評(píng)標(biāo)準(zhǔn)和 技術(shù)要求��,顯著降低細(xì)胞和基因治療產(chǎn)品的研發(fā)成 本�����。 建議我國(guó)參照美國(guó) FDA 的實(shí)踐經(jīng)驗(yàn)��,在國(guó)家戰(zhàn) 略層面形成長(zhǎng)期工作計(jì)劃��,完善相關(guān)前沿技術(shù)法律 法規(guī)依據(jù)和指南框架��,引導(dǎo)前沿技術(shù)健康發(fā)展�����。
4.2 構(gòu)建監(jiān)管機(jī)構(gòu)與產(chǎn)學(xué)研管組織更廣泛的溝通協(xié)作平臺(tái)
目前我國(guó)藥品監(jiān)管過程缺少產(chǎn)學(xué)研用平臺(tái)的聯(lián) 合參與����,雖在持續(xù)關(guān)注相關(guān)技術(shù)方法的研發(fā)進(jìn)展,但 研發(fā)和監(jiān)管尚未形成良性溝通����,研發(fā)和驗(yàn)證、具體應(yīng) 用脫節(jié),不利于相關(guān)技術(shù)方法在具體藥物研發(fā)中的 轉(zhuǎn)化落地��。 建議將產(chǎn)��、學(xué)��、研����、管有機(jī)結(jié)合,建立良好 的溝通機(jī)制��,在溝通中形成共識(shí)�����,在共識(shí)中促進(jìn)轉(zhuǎn) 化��,強(qiáng)化醫(yī)保�����、衛(wèi)健�����、藥監(jiān)等政府部門、產(chǎn)業(yè)界��、臨床 研究機(jī)構(gòu)與前沿技術(shù)之間的互信互認(rèn)����,打通技術(shù)向 應(yīng)用實(shí)踐轉(zhuǎn)化的壁壘��。
4.3 深度參與藥品監(jiān)管國(guó)際交流與合作
作為 ICH 成員國(guó)����,我國(guó)應(yīng)通過拓展雙邊、多邊 合作交流合作機(jī)制�����,積極參與全球藥品監(jiān)管研究����,主 動(dòng)參與國(guó)際規(guī)則制定,及時(shí)掌握創(chuàng)新藥物研發(fā)的新 趨勢(shì)��、新問題[12] �����。 2023 年 6 月 ICH 溫哥華會(huì)議正 式通過了美國(guó)生物技術(shù)創(chuàng)新協(xié)會(huì)(BIO)提交的《關(guān)于細(xì)胞和基因治療產(chǎn)品監(jiān)管國(guó)際協(xié)調(diào)戰(zhàn)略路徑的建 議書》,標(biāo)志著 RMAT 作為全球生物醫(yī)藥未來發(fā)展 的重點(diǎn)方向����,將是 ICH 等國(guó)際藥品研發(fā)和監(jiān)管標(biāo)準(zhǔn) 協(xié)調(diào)的重點(diǎn)領(lǐng)域之一。 我國(guó)已通過“ 中國(guó)藥品監(jiān)管 科學(xué)行動(dòng)計(jì)劃” 先后制定了 15 項(xiàng)細(xì)胞和基因治療 產(chǎn)品相關(guān)技術(shù)指導(dǎo)原則����,基本與國(guó)際同步,應(yīng)當(dāng)把握 當(dāng)前的技術(shù)前沿優(yōu)勢(shì)����。 增強(qiáng)政策支持和資源配置, 定期研究調(diào)整或增加“中國(guó)藥品監(jiān)管科學(xué)行動(dòng)計(jì) 劃”研究項(xiàng)目和藥品監(jiān)管科學(xué)全國(guó)重點(diǎn)實(shí)驗(yàn)室任務(wù) 清單����,提高監(jiān)管科學(xué)研究的針對(duì)性,提升國(guó)際話語權(quán) 和前沿技術(shù)協(xié)調(diào)主動(dòng)權(quán)����。
5、 結(jié)語
面對(duì)創(chuàng)新技術(shù)的迅速發(fā)展����,藥品監(jiān)管機(jī)構(gòu)的能力要求已經(jīng)不是傳統(tǒng)意義上的知識(shí)緩慢積累,而是 爆炸式的知識(shí)涌入��。 為了彌補(bǔ)當(dāng)前國(guó)家醫(yī)藥創(chuàng)新轉(zhuǎn) 化鏈條明顯的短板,我國(guó)應(yīng)集中優(yōu)勢(shì)資源����,在“ 中國(guó) 藥品監(jiān)管科學(xué)行動(dòng)計(jì)劃” 取得積極成效的基礎(chǔ)上, 在“ 十四五” 期間全面強(qiáng)化藥品監(jiān)管科學(xué)體系建設(shè)�����, 為中國(guó)式現(xiàn)代化藥品監(jiān)管實(shí)踐提供科技支撐和引 領(lǐng)����。 依托藥品監(jiān)管科學(xué)全國(guó)重點(diǎn)實(shí)驗(yàn)室��、監(jiān)管科學(xué) 行動(dòng)計(jì)劃等平臺(tái)�����,集中優(yōu)勢(shì)力量����,針對(duì)創(chuàng)新產(chǎn)品特 性、行業(yè)問題����、監(jiān)管需求�����,研究制定各類前沿技術(shù)評(píng) 價(jià)的新工具�����、新標(biāo)準(zhǔn)����、新方法����,在研發(fā)的全鏈條協(xié)助 企業(yè)規(guī)避風(fēng)險(xiǎn),實(shí)現(xiàn)順暢轉(zhuǎn)化�����,不斷提高創(chuàng)新藥物的 成藥性評(píng)價(jià)水平����,最終帶動(dòng)整個(gè)產(chǎn)業(yè)的高質(zhì)量發(fā)展, 保障和促進(jìn)公眾健康��。