摘 要 / Abstract
抗體偶聯(lián)藥物(ADC)和抗體介導(dǎo)的核素偶聯(lián)藥物(RDC)作為抗腫瘤靶向生物藥,已經(jīng)在過(guò)去的10年中引起了廣泛的關(guān)注�����。本文介紹靶向藥物的研發(fā)歷程以及ADC概況��,討論ADC開(kāi)發(fā)中的幾個(gè)關(guān)鍵問(wèn)題����,如開(kāi)發(fā)風(fēng)險(xiǎn)、臨床毒性�、內(nèi)吞與藥效以及結(jié)合位點(diǎn)屏障作用的影響等,并分析ADC的延伸——抗體介導(dǎo)的RDC的開(kāi)發(fā)進(jìn)展和獨(dú)特優(yōu)勢(shì)等�����,旨在通過(guò)分析與討論ADC和RDC在臨床上的應(yīng)用前景和改進(jìn)措施����,為靶向生物藥的發(fā)展提供創(chuàng)新方向。
Antibody drug conjugate(ADC) and antibody-mediated radiolabeled drug conjugate(RDC) have garnered widespread attention as targeted anti-tumor biopharmaceuticals over the past decade. This article introduces the development process of targeted drugs and provides an overview of ADC. It discusses several key issues in ADC development, such as development risks, clinical toxicity, internalization and efficacy, and the impact of binding site barrier effects. The article also analyzes the extension of ADC, including the development progress and unique advantages of antibody-mediated RDC. The aim is to provide innovative development directions for targeted biopharmaceuticals by analyzing and discussing the prospects and improvement measures of ADC and RDC in clinical applications.
關(guān) 鍵 詞 / Key words
靶向藥物�;抗體偶聯(lián)藥物;核素偶聯(lián)藥物�;關(guān)鍵風(fēng)險(xiǎn);臨床毒性���;內(nèi)吞與藥效�;結(jié)合位點(diǎn)屏障
targeted drugs; antibody drug conjugate; radiolabeled drug conjugate; key risks; clinical toxicity; internalization and efficacy; binding site barrier
抗體偶聯(lián)藥物(antibody drug conjugate,ADC)及核素偶聯(lián)藥物(radionuclide drug conjugate,RDC)是近年來(lái)獲得廣泛關(guān)注的靶向生物藥�����,在抗腫瘤治療中展現(xiàn)了較高的應(yīng)用價(jià)值。此類藥物將細(xì)胞表達(dá)的生物大分子與小分子毒素或者放射性核素通過(guò)連接子偶聯(lián)成為新藥��。ADC及RDC的結(jié)構(gòu)相對(duì)復(fù)雜���,其體內(nèi)安全性及有效性的影響因素也不同于一般抗體或化藥分子��。本文討論了ADC的開(kāi)發(fā)風(fēng)險(xiǎn)�����、臨床毒性�����、內(nèi)吞和藥效等�����,并分析了RDC的開(kāi)發(fā)進(jìn)展和獨(dú)特優(yōu)勢(shì)�����。
1��、靶向藥物的起源及靶向治療
自DNA雙螺旋結(jié)構(gòu)破解后��,近代生物學(xué)的發(fā)展在幾十年內(nèi)突飛猛進(jìn)����,跨入了分子時(shí)代�。隨著對(duì)疾病本質(zhì)的深入認(rèn)識(shí),在疾病干預(yù)及藥物研發(fā)上也不斷提高��,于20世紀(jì)末開(kāi)啟了針對(duì)疾病分子機(jī)制中相關(guān)靶點(diǎn)的靶向藥物研發(fā)����。
傳統(tǒng)藥物開(kāi)發(fā)的典型模式可以用針對(duì)傳染病的抗生素類藥物及針對(duì)癌癥的細(xì)胞毒類藥物為示范,都是利用體外細(xì)胞模型及體內(nèi)動(dòng)物疾病模型開(kāi)展自然界及人工合成物質(zhì)的有效性及安全性篩選����,有成藥潛力的物質(zhì)經(jīng)進(jìn)一步優(yōu)化并完成生產(chǎn)及制劑的工藝開(kāi)發(fā)和質(zhì)量保障等工作,經(jīng)藥品監(jiān)管部門(mén)的審評(píng)審批���,最終獲批上市用于疾病治療��。在這些傳統(tǒng)藥物的開(kāi)發(fā)過(guò)程中�,對(duì)藥物的作用靶點(diǎn)與機(jī)制知之甚少���,臨床應(yīng)用中常出現(xiàn)攻擊疾病細(xì)胞之外的部分正常細(xì)胞�,使得大部分藥物存在一定不良反應(yīng)。
分子靶向藥物是針對(duì)特定疾病中與疾病關(guān)系密切的靶點(diǎn)開(kāi)發(fā)的藥物����,與靶點(diǎn)有特異性作用關(guān)系,對(duì)靶點(diǎn)陽(yáng)性細(xì)胞產(chǎn)生作用�����,而對(duì)靶點(diǎn)陰性細(xì)胞不產(chǎn)生作用����,從而減少對(duì)正常組織和細(xì)胞的傷害。靶向藥物開(kāi)發(fā)首先在腫瘤藥物中獲得突破�����,1998年����,美國(guó)食品藥品監(jiān)督管理局(FDA)批準(zhǔn)了抗人表皮生長(zhǎng)因子受體-2(HER2)的人源化抗體trastuzumab(曲妥珠單抗,商品名Herceptin/赫賽?。蔀榈谝粋€(gè)獲準(zhǔn)上市的靶向大分子生物藥����。2001年��,另一個(gè)靶向藥物imatinib(伊馬替尼����,商品名Gleevec/格列衛(wèi))獲批上市�,成為首個(gè)獲批上市的靶向小分子化藥�����。伊馬替尼是一種酪氨酸激酶抑制劑����,靶向因染色體易位而形成的BCR/ABL融合基因,可阻斷相關(guān)蛋白激酶的作用而抑制腫瘤細(xì)胞中的信號(hào)傳導(dǎo)��,臨床用于治療慢性髓性白血?���。╟hronic myelogenous leukemia,CML)等癌癥。在伊馬替尼早期研發(fā)中���,結(jié)合對(duì)靶點(diǎn)激酶的特異性作用�,很快鎖定其分子結(jié)構(gòu)骨架并進(jìn)行構(gòu)效優(yōu)化,在CML患者來(lái)源的細(xì)胞及腫瘤模型中獲得明確的與靶點(diǎn)相關(guān)的體外和體內(nèi)療效�����,于1998年進(jìn)入臨床開(kāi)發(fā)����,1999年獲得快速通道(fasttrack)資質(zhì),2000年進(jìn)入關(guān)鍵臨床階段���,2001年成功獲批上市用于治療CML�。
曲妥珠單抗和伊馬替尼的相繼成功標(biāo)志著靶向藥物時(shí)代的開(kāi)啟�,引領(lǐng)藥物研發(fā)進(jìn)入深層次解讀疾病生物學(xué)本質(zhì)、開(kāi)拓及驗(yàn)證相關(guān)靶點(diǎn)����、開(kāi)展靶點(diǎn)聚焦的藥物研發(fā)及成藥性優(yōu)化等領(lǐng)域,同時(shí)推進(jìn)后續(xù)的工藝���、質(zhì)檢�����、臨床前和臨床開(kāi)發(fā)等�����。到2017年�,已有667個(gè)人類來(lái)源的獨(dú)特蛋白及189個(gè)致病微生物來(lái)源的病原蛋白成為藥物開(kāi)發(fā)的靶點(diǎn),其中部分靶點(diǎn)蛋白為G蛋白偶聯(lián)受體類(12%)�、離子通道類(19%)、蛋白激酶類(10%)等[1]���。這些靶點(diǎn)分布于不同人類疾病����,均已開(kāi)展了小分子化藥及大分子生物藥的研發(fā)工作����。在Santos的分析中����,人類來(lái)源的667個(gè)靶點(diǎn)有540個(gè)開(kāi)展了小分子藥物研發(fā),146個(gè)開(kāi)展了大分子藥物研發(fā)���,其中部分靶點(diǎn)同時(shí)進(jìn)行著小分子及大分子的藥物研發(fā)�����。針對(duì)人類靶點(diǎn)開(kāi)發(fā)成功的1104種藥物��,有999種小分子化藥以及105種大分子生物藥���。針對(duì)189個(gè)致病微生物來(lái)源的病原靶點(diǎn)���,大部分致力于小分子化藥的研發(fā),只有7個(gè)靶點(diǎn)開(kāi)展了大分子藥物的研發(fā)����。短短20年,靶向藥物的研發(fā)取得了亮眼的成績(jī)����。
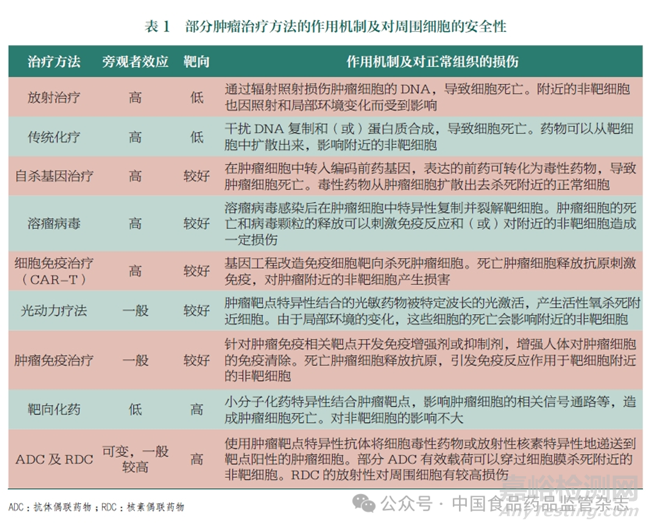
靶向藥物的開(kāi)發(fā)針對(duì)藥物對(duì)正常細(xì)胞的安全風(fēng)險(xiǎn)作出革命性的改變,加強(qiáng)了藥物對(duì)靶向細(xì)胞的選擇性���,提高了藥物的治療窗(therapeutic window)��。靶向治療策略在多種治療方法中的應(yīng)用及發(fā)展���,使醫(yī)療實(shí)現(xiàn)跨越式的進(jìn)步��。表1總結(jié)了部分腫瘤治療方法的作用機(jī)制及對(duì)周圍細(xì)胞的安全性��,分析了藥物對(duì)正常組織的損傷���。
2、ADC概況
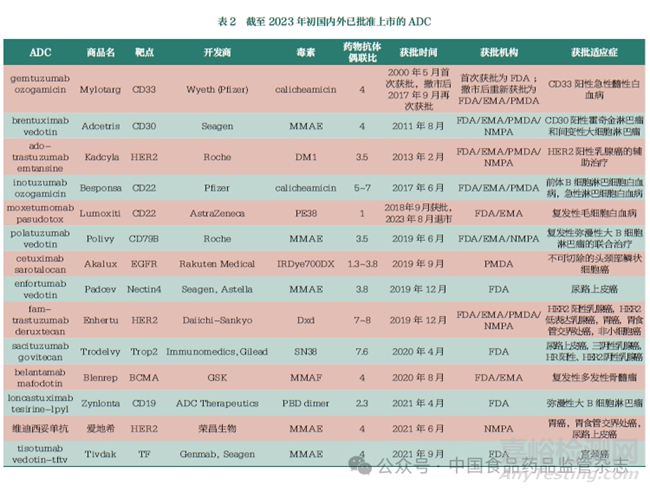
靶向治療藥物開(kāi)發(fā)中非常值得關(guān)注的是結(jié)合腫瘤靶點(diǎn)抗原的特異性抗體和小分子細(xì)胞毒性藥物共同組成的ADC����。ADC是一種靶向治療癌癥的藥物,它將特異性結(jié)合腫瘤抗原的抗體作為制導(dǎo)成分��,以殺死腫瘤細(xì)胞的強(qiáng)效毒素作為效應(yīng)彈頭��,組成高效靶向生物導(dǎo)彈��?�?贵w將彈頭直接引導(dǎo)到腫瘤細(xì)胞�,避免進(jìn)入非靶細(xì)胞以減少不良反應(yīng)��。至2023年����,已有超過(guò)15種ADC被批準(zhǔn)用于治療癌癥,還有多種ADC正在進(jìn)行臨床試驗(yàn)。表2是目前上市的ADC簡(jiǎn)介�。
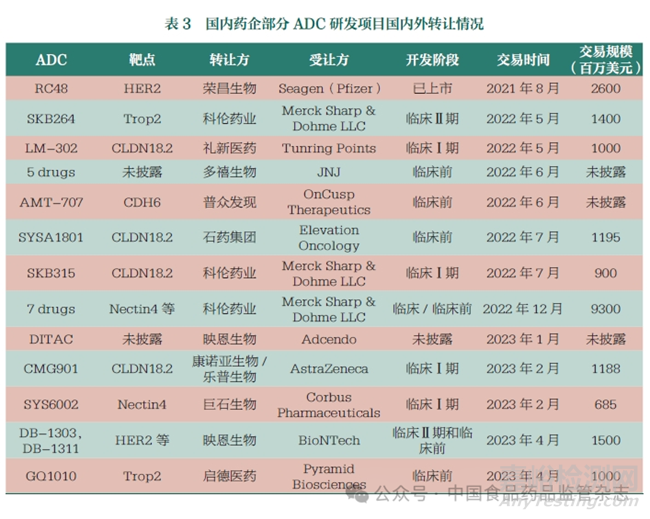
ADC近年來(lái)在我國(guó)的開(kāi)發(fā)異常火熱���。表3列出了部分有國(guó)內(nèi)外轉(zhuǎn)讓權(quán)益的ADC研發(fā)項(xiàng)目�����,可以客觀地反映國(guó)內(nèi)藥企在這一研發(fā)領(lǐng)域的生命力���,也體現(xiàn)了我國(guó)生物醫(yī)藥的創(chuàng)新活力和對(duì)世界新藥研發(fā)的重要貢獻(xiàn)。
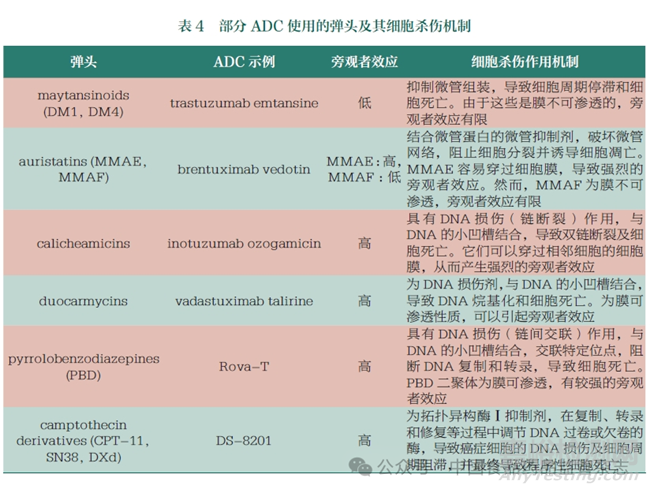
我國(guó)生物醫(yī)藥企業(yè)在ADC領(lǐng)域的快速發(fā)展是建立在近20年來(lái)主要由海歸力量衍生出的有強(qiáng)大生命活力的生物醫(yī)藥創(chuàng)新基礎(chǔ)之上的��。近年來(lái)的大規(guī)?��?贵w開(kāi)發(fā)與傳統(tǒng)的小分子細(xì)胞毒素的結(jié)合為國(guó)內(nèi)的創(chuàng)新生物醫(yī)藥企業(yè)展示了創(chuàng)新發(fā)展方向��,資本的涌入也促使藥企加入ADC賽道����。表3中轉(zhuǎn)讓項(xiàng)目的同質(zhì)化情況比較嚴(yán)重�,在新靶點(diǎn)及新機(jī)制方面仍相對(duì)薄弱。另外�����,使用小分子毒素作為細(xì)胞殺傷彈頭的選擇也較為局限。國(guó)內(nèi)的ADC開(kāi)發(fā)大多建立在這些常見(jiàn)毒素和細(xì)胞殺傷的機(jī)制上����,在新毒素和新機(jī)制應(yīng)用等方面還需要進(jìn)一步努力。表4列舉了目前部分已獲批或開(kāi)發(fā)的ADC相關(guān)毒素彈頭����。
3、ADC研發(fā)的關(guān)鍵因素
自ADC首次進(jìn)入臨床試驗(yàn)以來(lái)�,已有數(shù)百個(gè)ADC啟動(dòng)了千余項(xiàng)臨床試驗(yàn)。在此過(guò)程中���,多個(gè)ADC的臨床試驗(yàn)失敗或被從管線中移除���。縱觀ADC臨床試驗(yàn)失敗的原因�����,安全性和有效性是關(guān)鍵[2]�。
3.1 安全性因素
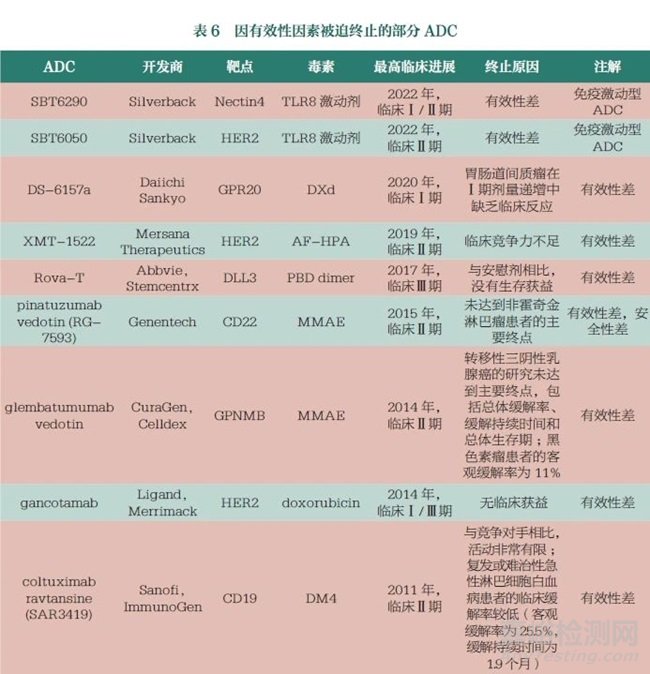
安全性原因主要指ADC自身的不可耐受的毒性限制了患者用藥����。其中ADC的靶向性和連接子毒素的穩(wěn)定性等對(duì)安全性影響較大�����,無(wú)論是靶向非腫瘤細(xì)胞還是毒素的非特異性釋放都會(huì)帶來(lái)嚴(yán)重的毒性問(wèn)題�。表5展示了部分因安全性因素被迫終止的ADC�。
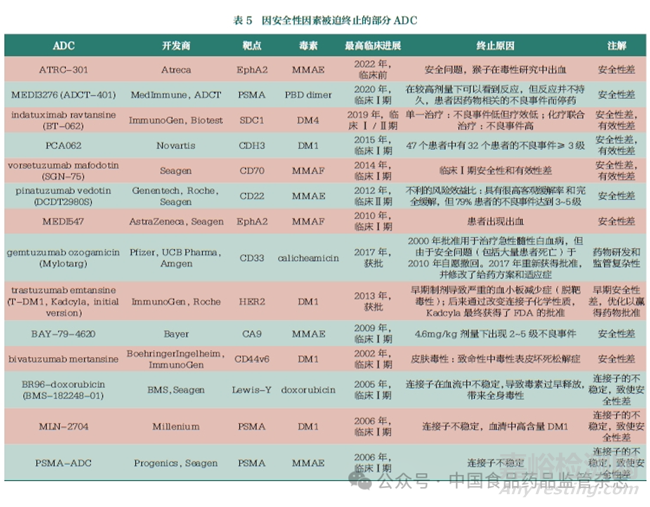
以靶向CD44v6的ADCbivatuzumab mertansine為例,其于2002年啟動(dòng)Ⅰ期臨床試驗(yàn)��,但隨后患者出現(xiàn)了嚴(yán)重甚至危及生命的皮膚脫落不良反應(yīng)�,進(jìn)而被迫終止臨床試驗(yàn)。分析其終止的主要原因在于CD44v6除在腫瘤細(xì)胞表達(dá)外�����,還在人體皮膚表面角質(zhì)層上表達(dá)��,造成的非瘤靶向結(jié)合(off-tumor/on-target)是致使bivatuzumab mertansine試驗(yàn)失敗的主要原因�。ATRC-301為靶向EphA2的ADC,該藥物在非人靈長(zhǎng)類動(dòng)物毒性研究中出現(xiàn)出血等安全隱患�,最終被Atreca放棄。MEDI547同樣為靶向EphA2的ADC��,也是因?yàn)榉橇霭邢蚪Y(jié)合致使患者用藥后出現(xiàn)了出血�、凝血�、皮膚和神經(jīng)系統(tǒng)等方面的反應(yīng)�,最終被迫終止試驗(yàn)。Mylotarg為全球第一個(gè)上市的ADC��,靶向CD33����,用于治療急性髓性白血病。Mylotarg曾在2010年因?yàn)閲?yán)重的致命性肝損傷等安全性問(wèn)題����,死亡率遠(yuǎn)高于對(duì)照組而被迫撤市;后來(lái)在2017年調(diào)整了相應(yīng)的給藥方案�����,以降低劑量的方式被FDA批準(zhǔn)重新上市�����。Mylotarg采用的連接子是含腙鍵和二硫鍵的化合物��,分析其中間撤市的主要原因在于該連接子不穩(wěn)定��,進(jìn)而致使Mylotarg在還未到達(dá)靶細(xì)胞時(shí),細(xì)胞毒素藥物卡奇霉素過(guò)早釋放���,引發(fā)了致命的毒性,而被迫退市��。Trop2表達(dá)于皮膚等正常的上皮細(xì)胞�����,靶向Trop2的ADC PF-06664178在臨床試驗(yàn)階段出現(xiàn)皮疹等嚴(yán)重的皮膚毒性�,被迫終止臨床。Trodelvy同樣為靶向Trop2的ADC����,其臨床階段的皮膚毒性要明顯低于PF-06664178,已于2020年獲批上市�����。同樣靶向Trop2,PF-06664178搭載的載荷是微管抑制劑auristatin,Trodelvy搭載的載荷是拓?fù)洚悩?gòu)酶I抑制劑���,因此對(duì)于Trop2這類表達(dá)較為廣泛的抗原����,載荷的選擇也是影響ADC安全性和臨床成敗的關(guān)鍵�����。
3.2 有效性因素
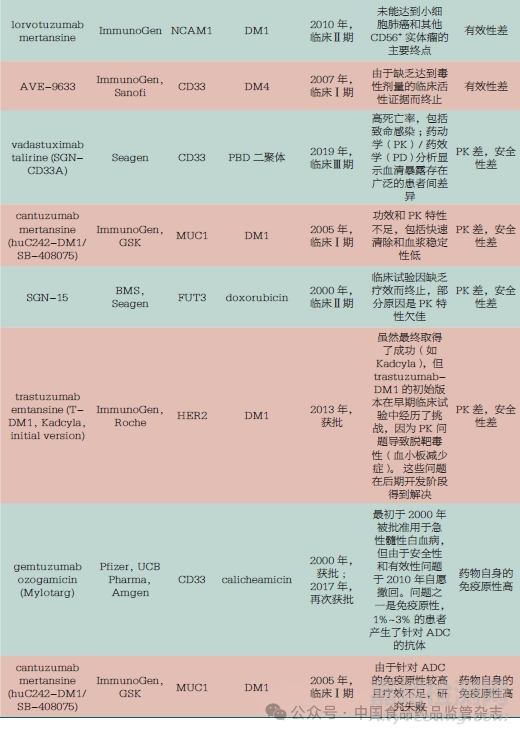
有效性方面主要指ADC的療效不足,其帶來(lái)的治療益處不優(yōu)于當(dāng)前的標(biāo)準(zhǔn)治療�。ADC的藥動(dòng)學(xué)(pharmacokinetics,PK)特性差或免疫原性高可導(dǎo)致ADC體內(nèi)清除迅速,另外ADC的有效載荷效力不足�、有效載荷腫瘤內(nèi)釋放不完全、靶點(diǎn)抗原密度低��、ADC的內(nèi)化特性差等都是制約ADC有效性的潛在因素�����。表6展示了因有效性因素被迫終止的部分ADC�。
以靶向HER2的SBT6050為例,這是偶聯(lián)了TLR8的免疫激動(dòng)型ADC�����,意在激活人體免疫系統(tǒng)起到殺傷腫瘤的效果�。從其I/Ib期臨床試驗(yàn)的中期數(shù)據(jù)來(lái)看,客觀緩解率僅為7%�����,分析其失敗的主要原因在于載荷TLR8的效力不足。Silverback研發(fā)的靶向Nectin4的SBT6290亦是如此�,表明這一策略存在機(jī)制缺陷。百奧泰公司開(kāi)發(fā)的BAT8001和BAT8003均使用美登素衍生物作為毒素載荷���,這兩款A(yù)DC相繼失敗和終止臨床的原因可能也是在于美登素衍生物的效力不足����。PK和免疫原性等特性一定程度上決定了ADC的循環(huán)半衰期和體內(nèi)清除速度����,從而也一定程度上影響了ADC的有效性�。MEDI4267(HER2靶向)在最大耐受劑量下的半衰期很短,極差的PK使得其在體內(nèi)被很快清除���,進(jìn)而制約了其臨床療效����。SGN-15(FUT3靶向)和cantuzumab mertansine(MUC1靶向)等多個(gè)ADC也是由于PK較差或免疫原性較高等問(wèn)題導(dǎo)致臨床有效性低而被迫終止�。此外,rovalpituzumab tesirine(DLL3靶向)�、depatuxizumab mafodotin(EGFR VIII靶向)、AGS16F(ENPP3靶向)以及ifastuzumab vedotin(Na Pi2b靶向)等ADC也因未能證明優(yōu)于標(biāo)準(zhǔn)治療而被終止��。
3.3 商業(yè)化因素
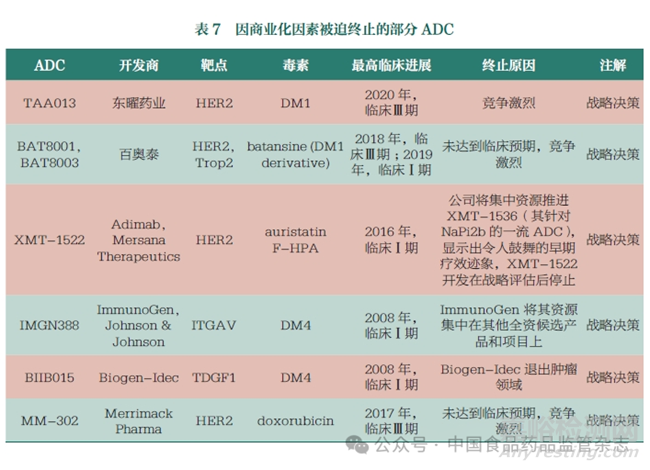
除安全性和有效性之外,商業(yè)化因素也左右著ADC的臨床走向��。表7展示了因商業(yè)化因素被迫終止的部分ADC�����,這些均與市場(chǎng)競(jìng)爭(zhēng)環(huán)境及企業(yè)的戰(zhàn)略調(diào)整等原因相關(guān)����。
4、ADC的臨床毒性
ADC具有特殊的分子構(gòu)造���,分別由抗體�、連接子和毒素彈頭組成�。在發(fā)揮功能時(shí),由具有靶向作用的抗體分子將具有殺傷作用的有效載荷靶向運(yùn)輸?shù)侥[瘤部位殺傷腫瘤�。與單獨(dú)的小分子給藥相比,ADC由于靶向性優(yōu)勢(shì)可以有效提高藥物的治療效果���。目前已上市的ADC也確實(shí)展現(xiàn)出了更優(yōu)的治療效果��。但是���,ADC的毒性依然是不容忽視的問(wèn)題���。如前文所述,目前有大量的ADC由于安全性問(wèn)題在臨床前和臨床階段被終止���,并且FDA已批準(zhǔn)上市的12個(gè)ADC中有10個(gè)被添加了黑框警告����。目前的研究表明����,ADC的主要毒性來(lái)自毒素部分�。Colombo等[3]將ADC和小分子單藥的劑量進(jìn)行標(biāo)準(zhǔn)化處理后發(fā)現(xiàn),ADC的最大耐受量與相應(yīng)小分子毒素之間并無(wú)顯著差異��。這主要是由于在實(shí)際給藥時(shí)��,通常只有1%的ADC可以達(dá)到腫瘤部位��,而大部分ADC仍停留在血液循環(huán)中或分布到其他正常組織中�����。ADC的非特異性攝取和裂解是其毒性產(chǎn)生的關(guān)鍵����。除毒素載荷之外���,不同靶點(diǎn)的組織分布和抗體分子的選擇也是ADC產(chǎn)生毒性的重要因素,可以造成藥物的非瘤靶向結(jié)合等�。為了更清晰地對(duì)比不同因素在ADC毒性中的作用,本文分別匯總并分析了偶聯(lián)MMAE的ADC和靶向HER2的ADC的不良反應(yīng)。
4.1 偶聯(lián)MMAE的ADC不良反應(yīng)比較
目前研究認(rèn)為���,ADC中的小分子毒素是產(chǎn)生機(jī)體毒性的主要原因,在臨床中現(xiàn)有ADC所展現(xiàn)出的不良反應(yīng)大多與這些毒素的表現(xiàn)相似���。其中非瘤非靶結(jié)合是最關(guān)鍵的因素之一����,主要表現(xiàn)為以下幾點(diǎn):(1)由于連接子穩(wěn)定性導(dǎo)致的脫靶�����,即有效載荷在血液循環(huán)中的過(guò)早釋放���,釋放的有效載荷會(huì)進(jìn)入正常細(xì)胞造成細(xì)胞殺傷�。(2)由于ADC疏水性及電荷導(dǎo)致的非特異性內(nèi)吞����,通常疏水性和正電性更高的ADC會(huì)導(dǎo)致更強(qiáng)的非特異性內(nèi)吞���。(3) ADC分子與免疫細(xì)胞表面Fcγ受體結(jié)合導(dǎo)致的內(nèi)吞,從而出現(xiàn)免疫細(xì)胞毒性����。以MMAE為例,表8匯總了5個(gè)以MMAE為有效載荷的ADC和MMAE分子的不良反應(yīng)���。從表8可以看出�����,5個(gè)ADC均表現(xiàn)出了血液毒性(嗜中性粒細(xì)胞減少、血小板減少等)及神經(jīng)系統(tǒng)毒性等���,這些不良反應(yīng)均與MMAE的毒性表現(xiàn)相同�����。
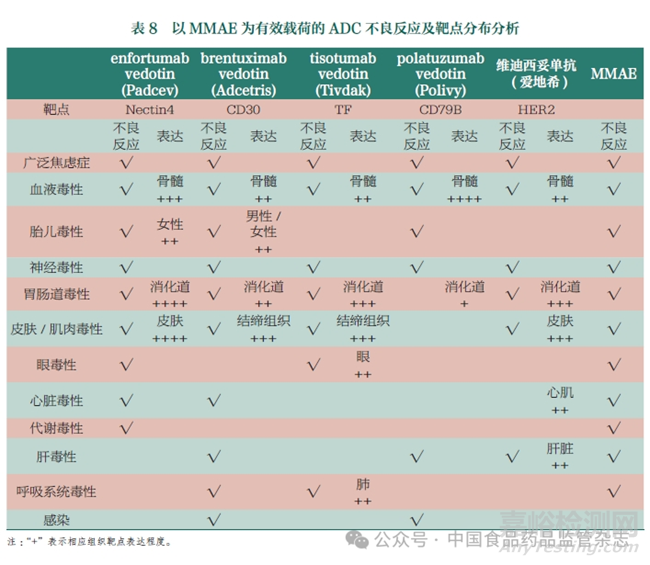
非瘤靶向結(jié)合也是ADC毒性產(chǎn)生的關(guān)鍵因素之一��,這主要是由于腫瘤相關(guān)靶點(diǎn)的組織分布不同導(dǎo)致的�。同樣對(duì)5個(gè)以MMAE作為有效載荷、靶向不同靶點(diǎn)的ADC的不良反應(yīng)進(jìn)行對(duì)比����。從表8可以看出,除了MMAE導(dǎo)致的血液/神經(jīng)毒性以外�,這5種ADC的毒性反應(yīng)存在顯著差異,這一差異與不同靶點(diǎn)的組織分布存在一定的相關(guān)性�����。例如����,enfortumab vedotin、brentuxi m abvedotin���、tisotumab vedotin和維迪西妥單抗均表現(xiàn)出了胃腸道毒性和皮膚/肌肉毒性���。對(duì)靶點(diǎn)分布進(jìn)行調(diào)研可以發(fā)現(xiàn),Nectin4�、CD30、TF和HER2在消化道和皮膚/結(jié)締組織中均具有較高的表達(dá)����。相對(duì)應(yīng)的�,CD79B在皮膚/結(jié)締組織中不表達(dá)�、在消化道中低表達(dá)。因此�,盡管MMAE具有腸道毒性和皮膚/肌肉毒性,最終靶向CD79B的polatuzumab vedotin也未表現(xiàn)出相關(guān)毒性�����。此外��,enfortumab vedotin和brentuximab vedotin的胎兒毒性���、tisotumab vedotin的眼毒性和呼吸系統(tǒng)毒性也均與靶點(diǎn)分布具有相關(guān)性�。
綜上所述��,盡管大部分情況下有效載荷本身的毒性決定了ADC的毒性�,但是不同靶點(diǎn)的選擇同樣也會(huì)對(duì)ADC的毒性產(chǎn)生影響。因此���,尋找腫瘤分布特異性更高的靶點(diǎn)也是降低ADC毒性的關(guān)鍵解決方案之一。
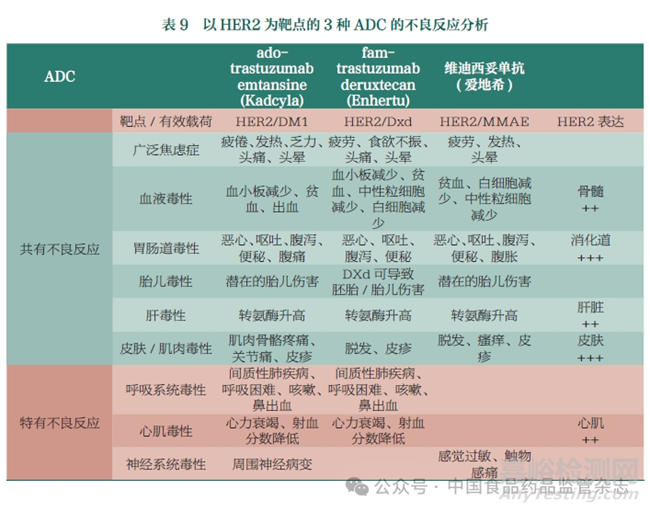
4.2 靶向HER2的ADC不良反應(yīng)比較
除了有效載荷和靶點(diǎn)選擇之外��,抗體分子自身的性質(zhì)是否會(huì)對(duì)ADC的毒性產(chǎn)生影響���?為了回答這一問(wèn)題����,本文對(duì)已上市的3個(gè)HER2靶向的ADC的不良反應(yīng)進(jìn)行了比較(表9)。其中ado-trastuzumab emtansine和fa -trastuzu mabderuxtecan使用的為同一個(gè)抗體分子�,即trastuzumab,兩者分別偶聯(lián)了DM1和Dxd作為有效載荷�����。維迪西妥單抗則是不同的HER2靶向抗體����,并偶聯(lián)了MMAE作為有效載荷。從表9可以看出��,盡管3個(gè)ADC由于HER2組織分布等因素導(dǎo)致了相似的血液毒性��、胃腸道毒性�����、肝毒性和皮膚/肌肉毒性����,但是在呼吸系統(tǒng)毒性和心肌毒性上產(chǎn)生了較大的差異�����。adotrastuzumab emtansine和fam-trastuzumab deruxtecan均表現(xiàn)出了間質(zhì)性肺疾病和心力衰竭等呼吸系統(tǒng)毒性或心肌毒性��,這一表現(xiàn)與trastuzumab單藥的表現(xiàn)基本相同�。相對(duì)應(yīng)的���,盡管HER2分子在心肌細(xì)胞中低表達(dá)�����,并且呼吸系統(tǒng)毒性和心肌毒性均屬于MMAE的毒性反應(yīng)(表8)�����,但是維迪西妥單抗卻并未表現(xiàn)出嚴(yán)重的呼吸系統(tǒng)毒性和心肌毒性�,以有效載荷自身的毒性和靶點(diǎn)分布等原因均無(wú)法完美解釋維迪西妥單抗的這一表現(xiàn)�。目前主流觀點(diǎn)認(rèn)為,靶向HER2的抗體藥物(trastuzumab,pertuzumab,ado-trastuzumab emtansine和fam-trastuzumab deruxtecan)的心肌毒性與HER2信號(hào)傳導(dǎo)通路被抑制有關(guān)�����,那么維迪西妥單抗表現(xiàn)出的心肌毒性差異則可能與其阻斷功能相關(guān)���,對(duì)此需要對(duì)這幾種抗體的生物學(xué)功能進(jìn)行深入研究和對(duì)比�。
綜上分析�,ADC毒性是抗體及連接子毒素的綜合表現(xiàn),其中小分子毒素的最大耐受量決定了ADC的體內(nèi)應(yīng)用情況���。對(duì)非瘤非靶結(jié)合及非瘤靶向結(jié)合等的綜合分析��,從平衡小分子毒素與抗體的結(jié)構(gòu)組成以及抗體優(yōu)化方面共同努力��,對(duì)于減少ADC的不良反應(yīng)將有所幫助��。
5���、ADC的內(nèi)吞與藥效
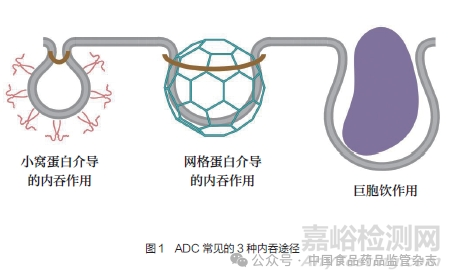
理想情況下,ADC在進(jìn)入靶細(xì)胞之前在血液循環(huán)中應(yīng)保持穩(wěn)定�����,但這一生物大分子不能通過(guò)被動(dòng)運(yùn)輸?shù)姆绞酵高^(guò)細(xì)胞膜進(jìn)入細(xì)胞�����,需要抗體與細(xì)胞膜上的靶點(diǎn)蛋白結(jié)合后再被內(nèi)吞進(jìn)入細(xì)胞。一般來(lái)說(shuō)抗體或ADC的內(nèi)吞過(guò)程可以分為以下3個(gè)階段:(1)抗體與細(xì)胞膜表面靶點(diǎn)抗原結(jié)合�����;(2)細(xì)胞膜內(nèi)陷形成內(nèi)吞囊泡����;(3)內(nèi)吞囊泡從細(xì)胞膜脫離進(jìn)入細(xì)胞質(zhì)(圖1)。由于存在多種內(nèi)吞途徑�,而且各途徑之間存在相互影響,因此內(nèi)吞過(guò)程是高度靈活且復(fù)雜的�����,這也導(dǎo)致了目前對(duì)于ADC內(nèi)吞作用與藥效之間的研究并不透徹[4]�����。
5.1 網(wǎng)格蛋白介導(dǎo)的內(nèi)吞作用
ADC與細(xì)胞膜表面的靶點(diǎn)蛋白結(jié)合形成復(fù)合物�,網(wǎng)格蛋白及一系列相關(guān)作用分子使細(xì)胞膜內(nèi)陷形成包被小窩,包被小窩在逐漸增大的同時(shí)被肌動(dòng)蛋白拉入細(xì)胞內(nèi)��,直至從細(xì)胞膜脫離下來(lái)形成網(wǎng)格蛋白包被囊泡����,然后通過(guò)核內(nèi)體-溶酶體途徑進(jìn)行胞內(nèi)代謝��。多種ADC被證實(shí)與網(wǎng)格蛋白共定位�,并且經(jīng)網(wǎng)格蛋白抑制劑處理后�����,ADC內(nèi)吞速率明顯被抑制����,因此該內(nèi)吞作用被證實(shí)為ADC內(nèi)吞的主要形式[5-6]���。網(wǎng)格蛋白由1條重鏈和1條輕鏈組成���,3個(gè)網(wǎng)格蛋白的重鏈和輕鏈形成1個(gè)“三棱體”,并與其他“三棱體”相互作用在包被小窩周圍形成一個(gè)多邊形晶格����。囊泡的形成需要50多種胞質(zhì)內(nèi)蛋白有序的參入。
5.2 小窩蛋白介導(dǎo)的內(nèi)吞作用
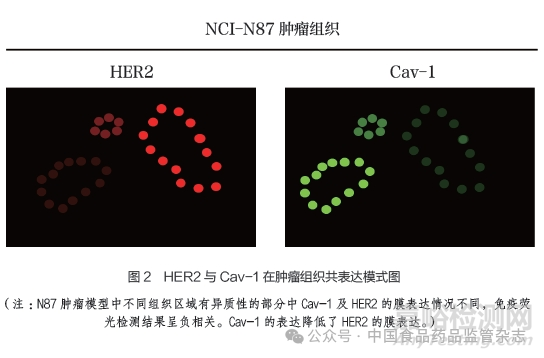
小窩是位于細(xì)胞表面的一種瓶狀的細(xì)胞質(zhì)膜凹陷結(jié)構(gòu)�,以較高水平的膽固醇和鞘糖脂為特征,存在于大多數(shù)細(xì)胞類型中����。部分ADC也可以通過(guò)該途徑進(jìn)入細(xì)胞�����,并轉(zhuǎn)運(yùn)至內(nèi)質(zhì)網(wǎng)和高爾基體內(nèi)���,但這兩種細(xì)胞器缺乏使ADC裂解的酶和環(huán)境因素,因此該內(nèi)吞途徑可能與ADC發(fā)揮藥效無(wú)關(guān)[7]�����。盡管小窩蛋白介導(dǎo)的內(nèi)吞與網(wǎng)格蛋白介導(dǎo)的內(nèi)吞都能使細(xì)胞膜發(fā)生凹陷��,但小窩蛋白形成的瓶狀凹陷在內(nèi)吞過(guò)程中的大小保持不變����,而且在各細(xì)胞株中的密度存在很大差異[8]。小窩蛋白1(Cav-1)是小窩的主要結(jié)構(gòu)蛋白��,除參與內(nèi)吞外���,還參與多種細(xì)胞活動(dòng)����,如細(xì)胞黏附��、信號(hào)轉(zhuǎn)導(dǎo)等。有研究表明���,小窩蛋白與耐藥機(jī)制有關(guān)�,例如Cav-1被發(fā)現(xiàn)可以負(fù)向調(diào)節(jié)HER2膜上表達(dá)影響抗HER2藥物的療效[9-10]�。
研究表明,Cav-1與HER2共定位于細(xì)胞膜凹陷結(jié)構(gòu)��,影響HER2在癌細(xì)胞膜上的表達(dá)和穩(wěn)定性�����。HER2的表達(dá)與Cav-1呈負(fù)相關(guān)�����,Cav-1高表達(dá)的細(xì)胞HER2膜表達(dá)低�,且傾向于胞內(nèi)積聚(圖2)�,在體內(nèi)腫瘤模型如NCI-N87異種移植瘤中可以發(fā)現(xiàn)這樣的負(fù)相關(guān)表達(dá)區(qū)域。在異質(zhì)性較高的患者腫瘤組織中���,Cav-1高表達(dá)造成腫瘤細(xì)胞膜HER2低表達(dá)���,會(huì)影響腫瘤組織對(duì)HER2藥物的臨床療效��。HER2和Cav-1的逆向調(diào)節(jié)及對(duì)抗HER2藥物的影響已在動(dòng)物實(shí)驗(yàn)中證明��,抗脂類代謝藥物lovastatin通過(guò)抑制Cav-1表達(dá)��,可以提高腫瘤細(xì)胞HER2的膜表達(dá)����,并進(jìn)而增強(qiáng)T-DM1的抗腫瘤藥效[9-10]��。以上發(fā)現(xiàn)具有一定的臨床指導(dǎo)意義����,或許可以通過(guò)與lovastatin等膽固醇類藥物聯(lián)用治療HER2低表達(dá)或T-DM1耐藥患者。
5.3 巨胞飲作用
部分ADC也可以通過(guò)巨胞飲作用進(jìn)入細(xì)胞內(nèi)��,該作用是細(xì)胞對(duì)胞外大分子物質(zhì)的一種非特異性的內(nèi)吞作用[11]�����。例如�,Jedema等[12]報(bào)道治療急性髓性白血病的ADC gemtuzumab ozogamicin(CD33靶向)存在2種內(nèi)吞途徑,在低濃度時(shí)通過(guò)與細(xì)胞表面的CD33結(jié)合啟動(dòng)網(wǎng)格蛋白介導(dǎo)的內(nèi)吞�����,而在高濃度時(shí)通過(guò)巨胞飲作用進(jìn)入CD33陰性細(xì)胞,這2種內(nèi)吞途徑都能釋放有效載荷阻斷DNA復(fù)制���,影響細(xì)胞周期����,導(dǎo)致細(xì)胞死亡�。但巨胞飲作用作為一種非特異性的內(nèi)吞作用,往往與不良反應(yīng)相關(guān)�,例如,臨床中觀察到的gemtuzumab ozogamicin肝毒性可能與肝臟中存在大量具有巨胞飲作用細(xì)胞有關(guān)�����。
ADC的內(nèi)吞與胞內(nèi)轉(zhuǎn)運(yùn)對(duì)療效及毒性有著顯著影響�����,提高內(nèi)吞速率�、加快ADC向核內(nèi)體-溶酶體聚集有可能提高ADC對(duì)腫瘤細(xì)胞的殺傷并減少毒性�。由于ADC開(kāi)發(fā)周期長(zhǎng)、耗費(fèi)大��,為了提高ADC開(kāi)發(fā)的成功率�����,需要謹(jǐn)慎選擇靶點(diǎn)、抗體結(jié)合表位及親和力�����、連接子的類型�、有效載荷,以上各項(xiàng)都有可能影響ADC的內(nèi)吞速率及胞內(nèi)轉(zhuǎn)運(yùn)��。在靶點(diǎn)蛋白的膜表達(dá)方面�,分析抗原低表達(dá)機(jī)制對(duì)提高藥效和解決耐藥等有重要意義。結(jié)合其他藥物可能導(dǎo)致的腫瘤抗原膜表達(dá)變化情況�,對(duì)不同患者群體開(kāi)展深入分層研究,可以進(jìn)一步提高對(duì)患者及ADC治療的認(rèn)識(shí)��,也為已上市的ADC與其他藥物聯(lián)用增強(qiáng)療效提供臨床用藥創(chuàng)新策略�����。
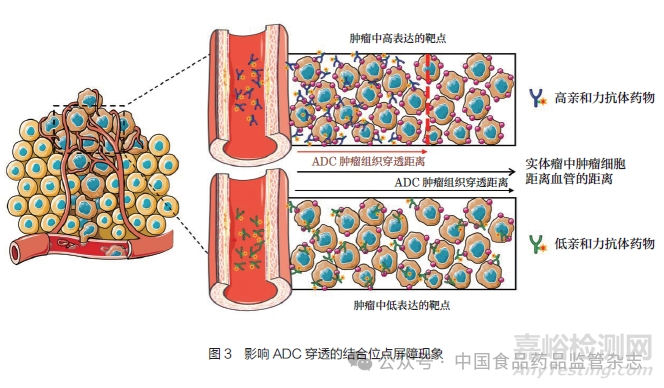
6���、結(jié)合位點(diǎn)屏障作用對(duì)ADC的影響
ADC作為一種靶向殺傷腫瘤的抗體藥物已越來(lái)越受到關(guān)注���,但是在臨床實(shí)踐中�,ADC的療效受到多種因素的制約�。例如,在實(shí)體腫瘤的治療中�����,ADC面臨的另一個(gè)難題就是藥物在腫瘤組織滲透中的結(jié)合位點(diǎn)屏障(binding-site barrier,BSB)��。當(dāng)ADC從血管進(jìn)入實(shí)體瘤組織中��,高親和力的抗體通常會(huì)緊密結(jié)合在血管周圍的實(shí)體瘤細(xì)胞����,難以向腫瘤內(nèi)部滲透,導(dǎo)致ADC僅對(duì)血管周圍表層細(xì)胞進(jìn)行殺傷而難以進(jìn)入實(shí)體瘤核心(圖3)�����,從而影響藥效[13-14]���。從圖3可以看出,抗體的親和力及靶點(diǎn)的分布情況均會(huì)影響ADC的腫瘤組織穿透����,因此在選擇ADC抗體時(shí)需要結(jié)合靶點(diǎn)的具體情況對(duì)抗體結(jié)合的親和動(dòng)力學(xué)作擇優(yōu)比較�,才能保證ADC有較好的腫瘤組織穿透能力和藥效�。理論上,增大給藥劑量在一定程度上能解決滲透性差的問(wèn)題��,但對(duì)于ADC來(lái)說(shuō)����,藥物毒性限制了給藥劑量,因此增大劑量的方法不適用�。這里主要介紹2種增強(qiáng)ADC腫瘤組織滲透的方法:ADC與其對(duì)應(yīng)的裸抗體聯(lián)合給藥[14];ADC與即時(shí)競(jìng)爭(zhēng)抑制性抗體聯(lián)合給藥[15]。
6.1 ADC與其對(duì)應(yīng)的裸抗體聯(lián)合給藥
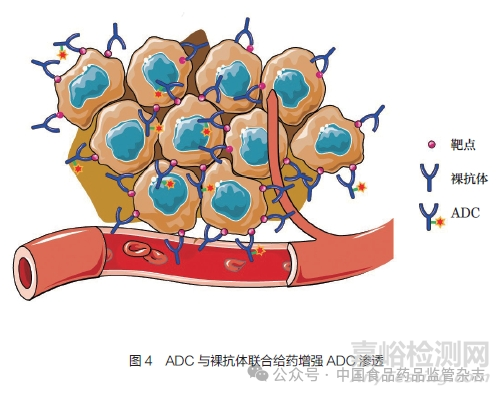
該方法采用毒素偶聯(lián)的抗體ADC和未偶聯(lián)的裸抗體聯(lián)合給藥���,混合物中裸抗體的含量為ADC的5~10倍�。高濃度的裸抗體分子競(jìng)爭(zhēng)結(jié)合實(shí)體瘤血管表層細(xì)胞較多��,促使ADC進(jìn)一步向?qū)嶓w瘤內(nèi)部滲透���。如圖4所示�����,靠近血管的腫瘤細(xì)胞上的靶點(diǎn)被ADC及裸抗體分別結(jié)合占位����,其中裸抗體結(jié)合優(yōu)勢(shì)較高,促使血管中ADC滲透進(jìn)入距血管距離更遠(yuǎn)的腫瘤組織��,實(shí)現(xiàn)與腫瘤內(nèi)部靶細(xì)胞的結(jié)合����。該方法能在不增加ADC給藥劑量和藥物毒性的前提下,有效增加ADC滲透能力以及最終藥效���。目前�,該方法在眾多抗體藥物�����、針對(duì)不同靶點(diǎn)的實(shí)體瘤臨床前實(shí)驗(yàn)中得到了驗(yàn)證���。例如�,抗表皮生長(zhǎng)因子受體(EGFR)抗體panitumumab偶聯(lián)熒光分子IRDye800CW模擬的ADC�,可以通過(guò)檢測(cè)熒光信號(hào)確定panitumumab-IRDye800CW的腫瘤組織分布情況�����。結(jié)果發(fā)現(xiàn),panitumumab-IRDye800CW單藥組熒光信號(hào)主要分布在實(shí)體瘤血管表層�����,而panitumumab和panitumumab-IRDye800CW混合組熒光信號(hào)分布更深入實(shí)體瘤內(nèi)部[14]�����。在T-DM1和trastuzumab聯(lián)用中也發(fā)現(xiàn)類似的實(shí)驗(yàn)結(jié)果[16]��。在此種聯(lián)合給藥策略下��,ADC/裸抗體混合物的滲透能力與ADC/裸抗體含量比例相關(guān)��。在ADC含量不變的條件下以及一定的劑量濃度范圍內(nèi)��,裸抗體的含量越高����,ADC的滲透能力越強(qiáng)[16-18]。雖然眾多臨床前研究表明該方法有助于增加ADC的滲透性以及藥效�����,但該方法的臨床有效性還需要進(jìn)一步證明�����,仍存在一系列的問(wèn)題需要解決,諸如ADC/裸抗體的混合比例����、靶細(xì)胞抗原豐度、裸抗體本身的不良反應(yīng)等�。
6.2 ADC與即時(shí)競(jìng)爭(zhēng)抑制性抗體聯(lián)合給藥
該方法的主要思路是開(kāi)發(fā)一個(gè)結(jié)合抗ADC的抗體結(jié)合域的抗體(anti-idiotypic antibody)。將該抗體與ADC混合使用��,抗ADC抗體與ADC的抗體結(jié)合域結(jié)合后��,降低ADC與抗原的結(jié)合���,避免ADC在實(shí)體瘤血管表面富集����、結(jié)合和內(nèi)吞�����,促進(jìn)復(fù)合物向?qū)嶓w瘤內(nèi)部滲透���。當(dāng)進(jìn)入實(shí)體瘤內(nèi)部后��,抗ADC抗體脫落����,解除對(duì)ADC抗體結(jié)合域的封閉并快速降解����,進(jìn)而釋放出可以結(jié)合靶點(diǎn)抗原的ADC,并發(fā)揮其腫瘤殺傷作用�。例如,Alvarez-Reuda等[19]開(kāi)發(fā)出抗trastuzumab的羊駝單域抗體1HE����。該抗體與trastuzumab聯(lián)合給藥時(shí)能有效降低trastuzumab與HER2的結(jié)合,同時(shí)還具備1HE半衰期短(1.2小時(shí))但1HE-trastuzumab復(fù)合物半衰期長(zhǎng)(56小時(shí))的特性�,有效地保證該復(fù)合物在血液、實(shí)體瘤環(huán)境中的穩(wěn)定性以及滲透實(shí)體瘤內(nèi)部1HE解離后的快速降解�����。1HE-T-DM1復(fù)合物在小鼠SK-OV3和NCI-N87移植瘤模型中的腫瘤滲透能力以及抗腫瘤藥效均強(qiáng)于T-DM1單獨(dú)給藥[15]����。
雖然上述方法在體外實(shí)驗(yàn)和小鼠體內(nèi)實(shí)驗(yàn)均展現(xiàn)出良好的增強(qiáng)ADC滲透效果和提高ADC藥效的作用,但相關(guān)人體試驗(yàn)還未完成��。上述聯(lián)合用藥以期提高ADC的臨床療效還有待于進(jìn)一步的臨床試驗(yàn)來(lái)驗(yàn)證。如何制定有效的臨床開(kāi)發(fā)策略還需要更多的臨床前研究提供支持���,同時(shí)需要謹(jǐn)慎的臨床方案設(shè)計(jì)�,并持續(xù)不斷地優(yōu)化方案來(lái)保證臨床試驗(yàn)的成功����。總的來(lái)說(shuō)���,開(kāi)發(fā)多元化策略提高ADC腫瘤組織穿透�、提高藥效�����、降低ADC毒性�����、建立有效臨床試驗(yàn)和治療方案是ADC開(kāi)發(fā)成功的重要保障����,如何讓ADC有更好的腫瘤組織穿透和殺傷是ADC設(shè)計(jì)及臨床應(yīng)用中需要不斷探索的。
7���、ADC的延伸——抗體介導(dǎo)的RDC
放射性RDC是含有放射性核素供醫(yī)學(xué)診斷和治療用的一類特殊藥物�。與ADC類似,在結(jié)構(gòu)上RDC主要由配體��、連接子�、螯合物和放射性同位素構(gòu)成�����。RDC與ADC的最大差異是藥物載荷����;RDC不再是小分子毒素,而是放射性核素��。使用不同的醫(yī)用核素����,可以開(kāi)展顯像或治療的不同功能,部分核素兼?zhèn)?種能力�。診斷用核素藥物用于獲得體內(nèi)靶器官或病變組織的影像或功能參數(shù),進(jìn)行疾病診斷����;治療用核素藥物將細(xì)胞毒性水平的放射性核素輸送到病變部位�,利用放射性同位素輻射產(chǎn)生局部電離輻射生物效應(yīng)�,對(duì)病變細(xì)胞或組織產(chǎn)生殺傷作用。
7.1 RDC開(kāi)發(fā)進(jìn)展
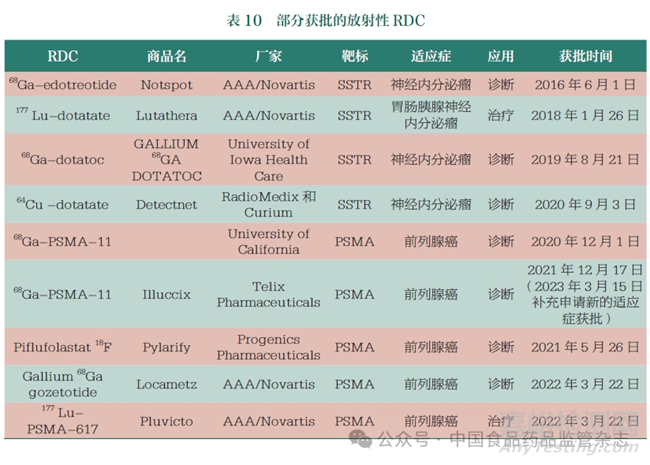
放射性藥物的開(kāi)發(fā)已有較長(zhǎng)歷史�����,早期獲批的放射性藥物大多是非靶向性藥物�,利用放射性同位素本身的體內(nèi)富集特點(diǎn)對(duì)患者進(jìn)行治療,同時(shí)會(huì)殺傷正常細(xì)胞����,不良反應(yīng)較大。能夠?qū)崿F(xiàn)對(duì)目標(biāo)病灶進(jìn)行靶向性殺傷的靶向放射性藥物研究���,近幾年也取得了較好的進(jìn)展(表10)�。其中�,Lutathera、Pluvicto等靶向放射性藥物的獲批�,極大地推動(dòng)了放射性藥物精準(zhǔn)靶向發(fā)展的步伐。
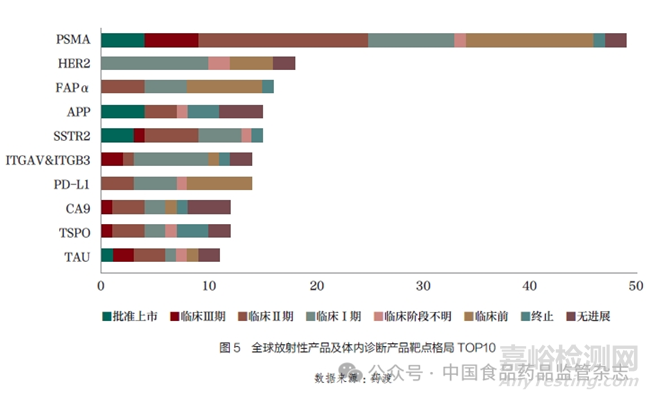
目前在研藥物靶點(diǎn)除了對(duì)標(biāo)SSTR和PSMA�,還有實(shí)體瘤常見(jiàn)靶點(diǎn)如PD-L1、HER2(圖5)�����,其中靶向PD-L1的RDC讓人非常感興趣。眾所周知�����,放療可以誘導(dǎo)免疫反應(yīng)��,促進(jìn)免疫細(xì)胞進(jìn)入腫瘤�����,但放療后腫瘤可能出現(xiàn)PD-L1上調(diào)而削弱抗腫瘤免疫����,因此靶向PD-L1的RDC或許能使免疫治療不敏感的“冷”腫瘤變“熱”��,前景讓人期待���。
7.2 RDC的獨(dú)特優(yōu)勢(shì)
RDC存在很多ADC無(wú)法比擬的優(yōu)勢(shì)�,近幾年成為腫瘤靶向治療領(lǐng)域的一顆“新星”�,不斷有傳統(tǒng)藥企和創(chuàng)新藥企進(jìn)入。
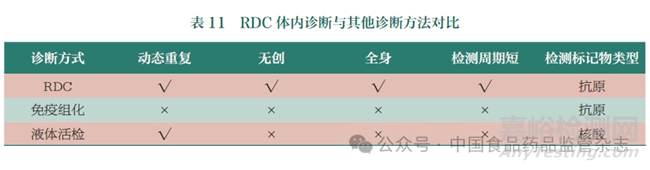
首先�,RDC是臨床實(shí)操中唯一能夠?qū)崿F(xiàn)診療一體化的藥物。RDC通過(guò)構(gòu)建適當(dāng)大小可以滿足組織穿透的靶向分子和裝載半衰期較短的同位素,藥物迅速?gòu)难哼M(jìn)入靶組織���,實(shí)現(xiàn)核素與原位腫瘤或繼發(fā)腫瘤相結(jié)合����,在極短半衰期內(nèi)給出信號(hào)��,并通過(guò)分子影像技術(shù)得出全面醫(yī)學(xué)影像結(jié)果��。其特點(diǎn)在于可以只更換核素部分��,相關(guān)的靶向分子和連接子都保持相似的情況下��,就能夠形成診療一體化的產(chǎn)品����,涵蓋診斷、分級(jí)與分期���、治療���、療效監(jiān)測(cè)及預(yù)后判斷等過(guò)程(表11)。例如����,Pluvicto可以先將診斷性同位素標(biāo)記在靶向分子和連接子構(gòu)成的前體上面����,然后用PET-CT進(jìn)行掃描�����,進(jìn)而確定患者病灶的位置�����,以及其目標(biāo)靶點(diǎn)的表達(dá)情況�����;接下來(lái)就可以用治療性的放射性核素連接成新的治療性RDC進(jìn)行治療��。治療后還可以再次用診斷性同位素標(biāo)記的診斷性RDC進(jìn)行檢測(cè)�,來(lái)觀察它的療效��,整個(gè)診斷治療的流程是精準(zhǔn)且一體化的��。梳理當(dāng)前國(guó)內(nèi)外RDC開(kāi)發(fā)管線��,診療一體化已經(jīng)成為開(kāi)發(fā)趨勢(shì),國(guó)外RDC研發(fā)的領(lǐng)先公司�,如ITM、Telix Pharmaceuticals�����、Clarity等�,均已在產(chǎn)品線布局中采用這一策略。
其次�,由于RDC作用機(jī)制不同,決定了它的治療優(yōu)勢(shì)�����。主要是基于核素的物理殺傷作用直接或者間接斷裂腫瘤細(xì)胞DNA�,達(dá)到殺傷腫瘤的目的。RDC只需要在目標(biāo)細(xì)胞附近有一定的滯留時(shí)間����,達(dá)到一定劑量的電離輻射就可以殺滅腫瘤細(xì)胞,而不需要內(nèi)吞進(jìn)腫瘤細(xì)胞內(nèi)����。因此,靶向載體的選擇范圍更為廣泛�����,小分子、多肽或者抗體藥物都可以成為RDC的靶向載體��。治療腫瘤時(shí)���,RDC除了直接損傷靶腫瘤細(xì)胞外���,還具有“交叉火力/旁殺效應(yīng)”,因此在擴(kuò)散性腫瘤中具有應(yīng)用潛力���。由于它的殺傷機(jī)制跟其他藥物不同���,所以即便在其他藥物無(wú)效或者耐藥的時(shí)候,RDC還是可以發(fā)揮治療作用�。例如�,已獲批的Pluvicto能夠使末線轉(zhuǎn)移性去勢(shì)抵抗性前列腺癌患者總生存期延長(zhǎng)4個(gè)月[20],成為前列腺癌現(xiàn)有治療方案的有力補(bǔ)充��。
7.3 核素標(biāo)記助力ADC開(kāi)發(fā)
在ADC早期發(fā)現(xiàn)階段��,采用放射性核素標(biāo)記技術(shù)��,可以清楚地跟蹤候選ADC分子在體內(nèi)的PK行為。對(duì)比不同標(biāo)記位置�、不同偶聯(lián)方式的體內(nèi)行為差異,有助于盡早篩選出符合預(yù)期的ADC分子��。有研究發(fā)現(xiàn)����,ADC連接子的化學(xué)性質(zhì)對(duì)其在臨床前動(dòng)物模型中的吸收、分布�����、代謝和排泄(ADME)特性有顯著影響�����,特別是對(duì)血漿PK及腫瘤和肝臟組織中觀察到的分解代謝物[21]����。還有研究證實(shí),ADC抗體偶聯(lián)比值(drug-antibody ratio,DAR)也會(huì)影響其在體內(nèi)的分布和代謝�,從而影響藥物抗腫瘤效果[22-23]。例如���,美登素類細(xì)胞毒素ADC在小鼠體內(nèi)的PK分析表明�����,DAR平均值在6以下時(shí)�,具有較好的PK清除速率;DAR平均值在9以上時(shí)�,清除很快。組織分布結(jié)果顯示����,DAR值較低的ADC組織分布特征與裸抗體相似,而DAR值超過(guò)9的ADC組織分布和裸抗體有明顯差異:給藥后小分子化合物在全血中的濃度迅速下降�,同時(shí)很快積聚在肝臟,隨后經(jīng)肝臟快速排出體外�,導(dǎo)致體內(nèi)暴露量降低,對(duì)應(yīng)藥效學(xué)表現(xiàn)不佳[23]�。
在ADC開(kāi)發(fā)中,非臨床研究過(guò)程中組織分布����、體內(nèi)代謝和排泄的研究存在諸多挑戰(zhàn),使用放射性核素標(biāo)記ADC進(jìn)行自顯影�,不僅可以了解整個(gè)組織的分布,而且可以區(qū)分組織內(nèi)各區(qū)域的濃度差異���,再分區(qū)域定量�����,尤其對(duì)于評(píng)估ADC在腫瘤組織的分布深度與藥效之間的量效關(guān)系具有傳統(tǒng)方法不可比擬的優(yōu)勢(shì)����。目前已上市的ADC����,如Adcetris、Padcev��、Kadcyla����、Polivy、Besponsa���、Enhertu��、Zynlonta��、Tivdak在臨床前階段均采用了低能量放射性核素(14C或3H)標(biāo)記小分子化合物��,完成了組織分布��、物質(zhì)平衡����、血漿蛋白結(jié)合率、代謝產(chǎn)物鑒定等實(shí)驗(yàn)���。其中Kadcyla�、Polivy����、Enhertu、Tivdak同時(shí)采用了放射性核素(125I��、111In��、3H�����、89Zr)標(biāo)記抗體�,對(duì)比僅標(biāo)記抗體或小分子化合物后呈現(xiàn)出組織分布差異。例如�����,Enhertu使用不同放射性核素分別標(biāo)記抗體和小分子毒素���,通過(guò)2種動(dòng)物的實(shí)驗(yàn)結(jié)果�,完整闡述ADC在體內(nèi)的PK過(guò)程:Enhertu注射進(jìn)入體內(nèi)后���,跟隨抗體驅(qū)動(dòng)分布至全身各處���,全血中濃度最高,并不在正常組織中滯留����。在體內(nèi)釋放出的Dxd基本不代謝,絕大部分以原型的形式經(jīng)膽汁流入腸腔�,最終從糞便排出體外[24]。顯而易見(jiàn)����,RDC技術(shù)對(duì)ADC的成藥性及優(yōu)化工作有重要意義。
ADC開(kāi)發(fā)是一個(gè)復(fù)雜����、漫長(zhǎng)、昂貴且容易出錯(cuò)的過(guò)程。在過(guò)去的幾十年里����,臨床開(kāi)發(fā)的復(fù)雜性和成本呈指數(shù)級(jí)增長(zhǎng),而成功率卻沒(méi)有相應(yīng)的提高���。美國(guó)FDA提出了“探索性研究用新藥(exploratory investigational new drug,e IND)”的概念�����,并于2006年發(fā)布了《探索性研究用新藥研究指南》(Exploratory IND Studies)�����,以e IND進(jìn)行的臨床試驗(yàn)又稱“0期臨床試驗(yàn)”�����。0期臨床允許對(duì)結(jié)構(gòu)相似的一組不超過(guò)5個(gè)化合物或劑型進(jìn)行“微劑量”研究��,同時(shí)獲得人體PK數(shù)據(jù)��,以及采用各種影像學(xué)研究手段獲得人體組織分布情況����,便于早期從一組候選分子中確定最有研發(fā)價(jià)值的一個(gè)先導(dǎo)化合物進(jìn)行I期臨床試驗(yàn)及后續(xù)的研發(fā)。放射性核素標(biāo)記和顯影技術(shù)在藥物0期臨床中發(fā)揮著重要的作用�。據(jù)報(bào)道,在116項(xiàng)已發(fā)表的0期臨床研究中�����,45%使用LC-MS/MS,29%使用AMS,23%使用PET技術(shù)[25]����。
RDC的快速發(fā)展����,擴(kuò)充了腫瘤標(biāo)準(zhǔn)治療失敗患者的治療方案,為患者帶來(lái)更多希望���。同時(shí)核素標(biāo)記RDC的開(kāi)拓也為ADC的開(kāi)發(fā)起到了積極的促進(jìn)作用���,可以預(yù)期ADC及RDC的相互支持對(duì)靶向生物藥的發(fā)展有戰(zhàn)略指導(dǎo)意義。當(dāng)然在RDC開(kāi)發(fā)中��,由于監(jiān)管狀態(tài)待完善�����,核素種類和資源的相對(duì)短缺,技術(shù)開(kāi)發(fā)的瓶頸����、人才短缺等問(wèn)題,存在不少發(fā)展局限性����,但也提示RDC開(kāi)發(fā)在我國(guó)有較大提升空間。近年來(lái)�,RDC開(kāi)發(fā)中的技術(shù)問(wèn)題,例如高親和力的小分子多肽篩選困難��、小分子RDC在體內(nèi)代謝過(guò)快�����、腫瘤部位滯留時(shí)間有限而影響療效�、經(jīng)驗(yàn)證的RDC靶標(biāo)相對(duì)有限等,都可以從ADC開(kāi)發(fā)的成功經(jīng)驗(yàn)中得到更多的資源和啟發(fā)���。
從對(duì)靶向生物藥ADC多方面的分析來(lái)看����,目前此類藥物的改進(jìn)�、治療方案的優(yōu)化等都有較大提高空間����,標(biāo)示著這一創(chuàng)新藥物領(lǐng)域的強(qiáng)大生命力���。本文中提到的抗體優(yōu)選���、毒素和連接子創(chuàng)新、ADC結(jié)構(gòu)優(yōu)化��、腫瘤組織穿透��、低靶點(diǎn)表達(dá)和耐藥�����、聯(lián)合治療策略等問(wèn)題還需要行業(yè)的不懈努力�����。RDC更是為ADC的延伸提供了相互支持的重要力量���,結(jié)合RDC在影像技術(shù)中強(qiáng)大功能,在早期臨床開(kāi)發(fā)中的0期臨床試驗(yàn)創(chuàng)新支持�,相信會(huì)對(duì)ADC和RDC靶向生物藥的發(fā)展起到巨大推動(dòng)作用�,也為靶向生物藥的發(fā)展和應(yīng)用開(kāi)拓更廣闊的空間����。