摘要
兒童藥品的供應(yīng)保障是兒童醫(yī)療保健的重要一環(huán)����,然而我國(guó)的兒童藥品的供應(yīng)保障卻不能滿(mǎn)足市場(chǎng)需求。國(guó)際上歐美等國(guó)二十多年來(lái)已經(jīng)建立了一套行之有效的兒童用藥法律法規(guī)及配套激勵(lì)措施����,極大的促進(jìn)了兒童醫(yī)療保健事業(yè)的發(fā)展��。本文通過(guò)對(duì)比分析歐美促進(jìn)兒科藥品研發(fā)的關(guān)鍵工具-兒科藥品研發(fā)計(jì)劃(PSP和PIP)的內(nèi)容及審評(píng)審批流程�����,建議監(jiān)管當(dāng)局在兒童藥品政策法規(guī)的規(guī)制定和調(diào)整時(shí)引入兒科研究計(jì)劃并設(shè)立兒科藥品審評(píng)委員會(huì)�����,從而引導(dǎo)我國(guó)制藥企業(yè)研發(fā)理念的轉(zhuǎn)變�����,在藥品研發(fā)早期就將兒童納入整體研發(fā)策略中。
關(guān)鍵詞:兒童用藥����;兒科藥品研究計(jì)劃(PSP);兒科藥品調(diào)查計(jì)劃(PIP)��;審評(píng)審批
引言
兒童醫(yī)療保健是兒童健康發(fā)展的重要保障[1]��,保障兒童用藥的安全性和可及性是確保兒童健康發(fā)展的重要一環(huán)��。ICH E11(R1)[2](用于兒科藥物的臨床研究)將兒童患者按年齡分為足月新生兒(0-27天)�����、嬰幼兒(28天-23個(gè)月)����、兒童(2-11周歲)、青少年(12到16-18周歲)四個(gè)亞群����。在我國(guó)兒童通常是指0-14歲的人群,巨大的人口基數(shù)[3]及高患病率和就診率[4]引爆兒童用藥市場(chǎng)需求�����。然而,我國(guó)兒童用藥的供給保障卻不容樂(lè)觀(guān)��,存在品種短缺(普藥多����,特藥少)、劑型與規(guī)格不匹配兒童生理需求等困境�����。相關(guān)法律法規(guī)指導(dǎo)原則不完善����、兒童藥品研發(fā)對(duì)輔料和劑型有特殊要求(如適口性)�����、不同年齡段兒童生理病理的差異和臨床試驗(yàn)在倫理及安全方面要求高以及兒童患者難招募易脫落等問(wèn)題導(dǎo)致兒童藥品研發(fā)成本高�����、周期長(zhǎng)[5]��,使許多有意愿生產(chǎn)兒童藥品的企業(yè)望而卻步�����,由此進(jìn)一步造成我國(guó)兒童用藥領(lǐng)域超說(shuō)明書(shū)用藥現(xiàn)象嚴(yán)重[1],給兒童的身心健康帶來(lái)極大風(fēng)險(xiǎn)�����。
兒童藥品市場(chǎng)相對(duì)于成人藥品市場(chǎng)來(lái)說(shuō)是“小市場(chǎng)”�����,由此導(dǎo)致兒童用藥的研發(fā)往往是成人藥品研發(fā)的附屬��,即兒童藥品的開(kāi)發(fā)一般是由成人藥品開(kāi)發(fā)所驅(qū)動(dòng)�����,而不是由兒童的健康需求驅(qū)動(dòng)�����。此外兒童藥品的研發(fā)不應(yīng)局限于兒童專(zhuān)用藥品�����,更多的是應(yīng)鼓勵(lì)滿(mǎn)足兒童用藥需求產(chǎn)品的兒科研究��,最終完善藥品說(shuō)明書(shū)的兒童用藥信息,從而降低超說(shuō)明書(shū)用藥的風(fēng)險(xiǎn)�����,保障兒童身心健康[6]��。
我國(guó)針對(duì)兒童用藥的政策起步較晚�����,目前還沒(méi)有針對(duì)兒童用藥的專(zhuān)門(mén)立法��,相關(guān)法律法規(guī)還停留在部門(mén)層面��,不具備強(qiáng)制執(zhí)行力��,系統(tǒng)解決兒童用藥的安全可及問(wèn)題亟需上位法的建立健全����。相比之下��,國(guó)際上歐美等國(guó)對(duì)兒童用藥的立法工作已積累了二十多年的經(jīng)驗(yàn)����,逐步建立了一套行之有效的兒童用藥法律法規(guī)及配套激勵(lì)措施[1]��,極大的促進(jìn)了兒童醫(yī)療保健事業(yè)的發(fā)展�����。
本文主要對(duì)歐美促進(jìn)兒科藥品研發(fā)的主要工具-兒科藥品研發(fā)計(jì)劃的內(nèi)容及審評(píng)審批流程進(jìn)行對(duì)比����,以期為監(jiān)管部門(mén)在制定促進(jìn)我國(guó)兒童藥品研發(fā)政策時(shí)提供借鑒����。
一、法規(guī)背景
美國(guó)通過(guò)《兒科研究平等法案》(Pediatric Research Equity Act �����,PREA)引入了兒科藥品研究計(jì)劃(Pediatric Study Plan����,PSP),通過(guò)《最佳兒童藥品法案》(Best Pharmaceuticals for Children�����,BPCA)引入了書(shū)面請(qǐng)求(Written Request����,WR)��,通過(guò)這兩個(gè)工具��,采用強(qiáng)制與激勵(lì)相結(jié)合的方法引導(dǎo)企業(yè)進(jìn)行兒科研發(fā)�����。歐盟通過(guò)兒科管理?xiàng)l例(Regulation(EC)No 1901/2006)引入兒科藥品調(diào)查計(jì)劃(Pediatric Investgation Plan��,PIP)��,采用強(qiáng)制與激勵(lì)加幫扶的方法引導(dǎo)企業(yè)進(jìn)行兒科研發(fā)�����。
1.1 美國(guó)
在美國(guó)兒童是指0-17歲的人群[7]��。美國(guó)主要通過(guò)BPCA和PREA兩大支柱性法案促進(jìn)兒科藥品的研究����。
BPCA于2002年1月4日正式生效實(shí)施����,允許美國(guó)食品藥品監(jiān)督管理局(Food and Drug Administration,F(xiàn)DA)對(duì)認(rèn)為可能產(chǎn)生兒科獲益藥品的申請(qǐng)人發(fā)放WR����,以請(qǐng)求申請(qǐng)人進(jìn)行兒科研究,WR中會(huì)列出擬研究的類(lèi)型��、適應(yīng)癥��、年齡組����、研究終點(diǎn)、評(píng)估時(shí)間�����、入組標(biāo)準(zhǔn)��、藥品信息�����、劑型和給藥途徑����、研究方案����、安全性信息����、統(tǒng)計(jì)信息、標(biāo)簽信息�����、報(bào)告格式及時(shí)間框架[8]����。WR為非強(qiáng)制性要求,但如果申請(qǐng)人嚴(yán)格按照WR在規(guī)定的時(shí)間內(nèi)完成了研究并提交了報(bào)告�����,無(wú)論研究結(jié)果是否積極��,F(xiàn)DA都會(huì)授予申請(qǐng)人6個(gè)月的兒科獨(dú)占期(pediatric Exclusivity�����,PE),附加在申請(qǐng)人持有的含有相同活性成分的藥品的專(zhuān)利期或者市場(chǎng)獨(dú)占期后面����。申請(qǐng)人也可以提交擬議的兒科研究請(qǐng)求(Proposed Pediatric Study Request��,PPSR)����,主動(dòng)向FDA申請(qǐng)WR。
2003年12月3日����,美國(guó)國(guó)會(huì)通過(guò)PREA,除非FDA授予豁免(waiver)或者延遲(deferral)�����,否則要求特定藥品或者生物制品的申請(qǐng)或補(bǔ)充申請(qǐng)(如新活性成分����,新適應(yīng)癥、新劑型����、新給藥方案或新給藥途徑)進(jìn)行兒科評(píng)估(pediatric Assessment)[9]��,即評(píng)估藥品或生物制品在所有相關(guān)的兒科人群中所聲稱(chēng)適應(yīng)癥的安全性和有效性��,以支持在相應(yīng)兒科人群中給藥方案的安全和有效�����。PREA為強(qiáng)制性條款����,完成兒科評(píng)估是產(chǎn)品獲批上市的前提�����。PREA不具有激勵(lì)性�����,但PREA只針對(duì)產(chǎn)品所申請(qǐng)適應(yīng)癥在相關(guān)兒科人群中的安全和有效性評(píng)價(jià)�����;而WR主要針對(duì)該產(chǎn)品的活性成分��,可能要求在不同兒科人群中進(jìn)行多個(gè)適應(yīng)癥的研究�����。2012年的《FDA安全與創(chuàng)新法案》(FDASIA)對(duì)PREA進(jìn)行修訂,引入PSP��,要求特定申請(qǐng)的申請(qǐng)人在提交上市許可申請(qǐng)前需要提交初始PSP(Initial PSP�����,iPSP)并得到FDA對(duì)iPSP的同意����。
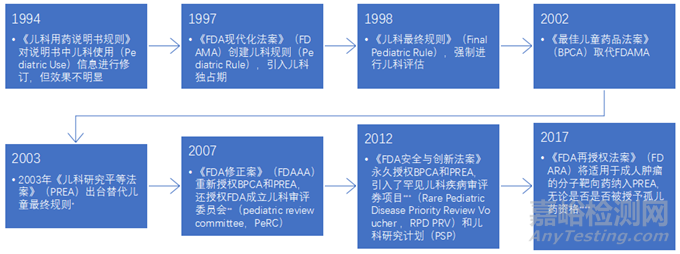
BPCA和PREA有日落條款�����,2007年被重新授權(quán)��,2012年被永久授權(quán)此后又經(jīng)歷一輪修訂(見(jiàn)圖1)��。
▲圖1-BPCA和PREA的立法沿革
*《兒科最終規(guī)則》于2002年末被哥倫比亞特區(qū)聯(lián)邦地方法院判定違憲��,禁止其實(shí)施[10]�����。PREA繼承了大部分《兒科最終規(guī)則》中的條款。
**FDA兒科審評(píng)委員會(huì)(PeRC)主要負(fù)責(zé)審評(píng)PSP�����、WR�����、PPSR及相關(guān)的豁免和延遲����,并向FDA提出建議,其建議沒(méi)有強(qiáng)制性��。
***兒科罕見(jiàn)疾病優(yōu)先審評(píng)券項(xiàng)目(RPD PRV)�����,授權(quán)FDA對(duì)符合相關(guān)兒科罕見(jiàn)疾病標(biāo)準(zhǔn)的申請(qǐng)授予RPD PRV����,該券可以轉(zhuǎn)讓?zhuān)梢允故褂迷撊臉?biāo)準(zhǔn)申請(qǐng)(300日)獲得優(yōu)先審評(píng)(180日)的資格。
****《FDA再授權(quán)法案》(FDAAA)實(shí)施前�����,被認(rèn)定為孤兒藥的產(chǎn)品被排除在PREA的覆蓋范圍外,即不用進(jìn)行兒科研究�����。
1.2 歐盟
歐盟對(duì)兒童的定義一般為小于18歲的人群[7]����。歐洲藥品評(píng)價(jià)局(EMEA) [2004年改名為歐洲藥品管理局(EMA)]自成立起(1995年)就非常重視兒童用藥的安全性和有效性。歐盟對(duì)兒童用藥的正式立法開(kāi)始于1997年歐盟委員會(huì)在EMA組織的一次專(zhuān)家圓桌會(huì)議��,2000年ICH E11發(fā)布��,EMA將其納入期監(jiān)管指南��。歐盟發(fā)布兒科監(jiān)管條例(Pediatric Regulation)公布于2006年6月1日并于2007年1月26日正式生效實(shí)施[11]����。該條例的目的是確保高質(zhì)量的兒童藥品研究����,確保兒童用藥品有適宜的劑型以及促進(jìn)高質(zhì)量的兒科用藥信息的可獲得性;同時(shí)避免兒童參加不必要的臨床試驗(yàn)和成人用藥品的批準(zhǔn)被延遲[12]�����。
在兒科用藥監(jiān)管條例的框架內(nèi):
引入了PIP,規(guī)定新藥上市授權(quán)申請(qǐng)(Maketing Authorisation Application��,MAA)以及新適應(yīng)癥����、新劑型、新給藥途徑的補(bǔ)充申請(qǐng)都要包含達(dá)成一致的PIP所涉及的兒科研究結(jié)果����,以支持該藥品在相關(guān)兒科人群中的授權(quán)和使用,除非該藥品被授予豁免或者延遲����。
創(chuàng)建了兒科委委員(Pediatric Committee,PDCO)負(fù)責(zé)對(duì)PIP的審評(píng)����,不同于FDA的PeRC,PDCO的審評(píng)意見(jiàn)EMA必須采納����。同F(xiàn)DA一樣,無(wú)論研究結(jié)果是否積極�����,申請(qǐng)人按照要求完成與PDCO達(dá)成一致的PIP的研究后,即可獲得6個(gè)月專(zhuān)利補(bǔ)充保護(hù)延長(zhǎng)�����,或者2年的市場(chǎng)獨(dú)占期延長(zhǎng)(孤兒藥)��。
此外該條例針對(duì)專(zhuān)利到期的藥品還提出了新的上市路徑-兒科使用上市許可(Pediatric Use Marketing Authorisation����,PUMA),并配備10年數(shù)據(jù)保護(hù)期的激勵(lì)措施��,但在EMA 2017年的報(bào)告中該條款的實(shí)施效果不佳[12]��。
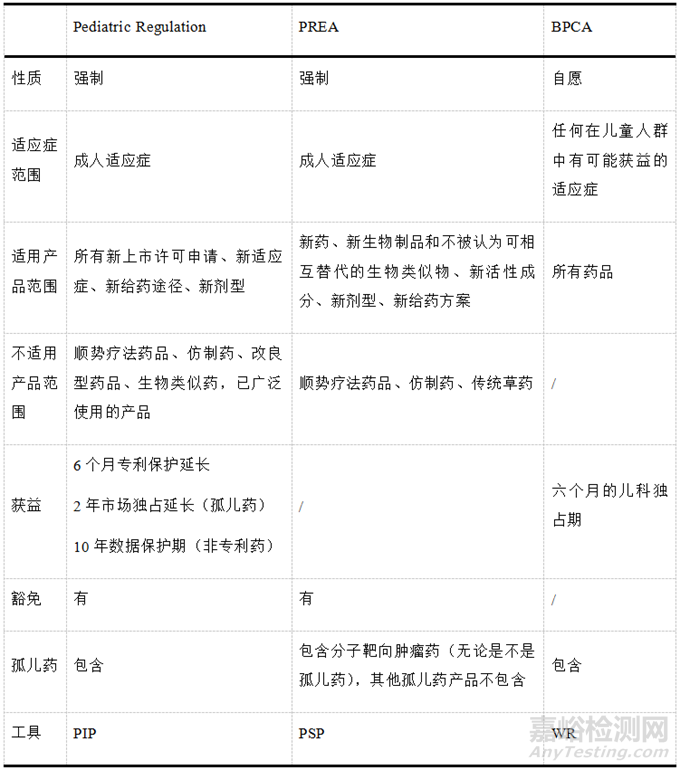
該條例還針對(duì)兒科用藥信息的公開(kāi)透明建立了泛歐洲兒科藥物研究和臨床試驗(yàn)中心網(wǎng)絡(luò)(Enpr-EMA)�����,并將研究結(jié)果錄入歐盟臨床研究數(shù)據(jù)庫(kù)(EudraCT)�����。還有一系列其他的幫扶措施如針對(duì)兒科研究中遇到的問(wèn)題可以免費(fèi)向EMA尋求科學(xué)建議或方案協(xié)助(針對(duì)孤兒藥)����。該條例與BPCA與PREA的異同見(jiàn)表1。
▲表1- BPCA��、PREA與Pediatric Regulation之間的異同
二����、歐美兒科藥品研發(fā)計(jì)劃內(nèi)容[13-16]
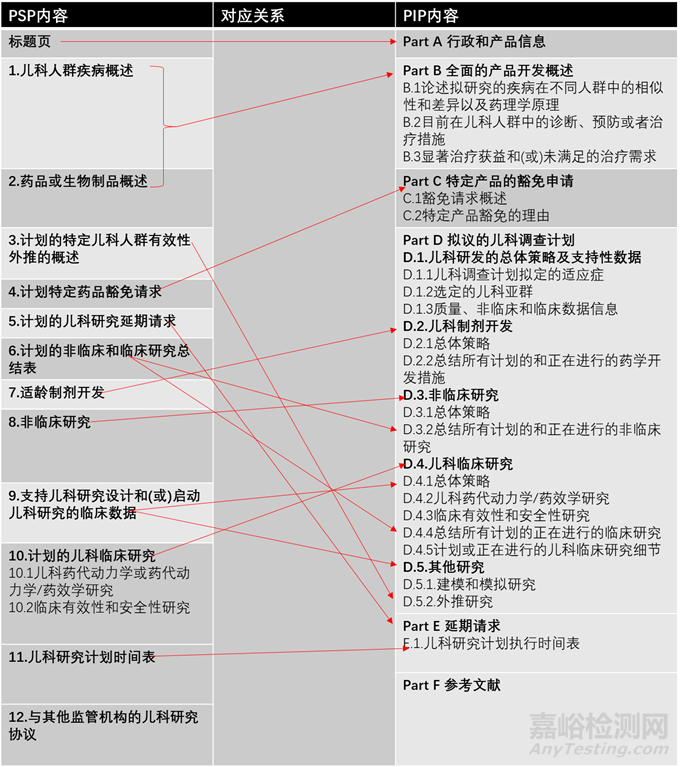
FDA和EMA都要求PSP和PIP是簡(jiǎn)潔獨(dú)立的文件,PSP一般為12-60頁(yè)��,PIP不超過(guò)40頁(yè)�����。PSP和PIP內(nèi)容上基本相似�����,但在層級(jí)結(jié)構(gòu)上有所不同(見(jiàn)圖2)��。
▲圖2-PSP和PIP內(nèi)容及其對(duì)應(yīng)關(guān)系
2.1 行政和產(chǎn)品信息
PSP要求在標(biāo)題頁(yè)中提供行政和產(chǎn)品信息����,包括藥品專(zhuān)利名和通用名、 NDA/BLA/IND號(hào)��、藥物類(lèi)別、適應(yīng)癥����、研究計(jì)劃簡(jiǎn)介等;相對(duì)于PSP�����,PIP的要求較多包括申請(qǐng)人信息����,活性成分名稱(chēng)、產(chǎn)品類(lèi)別��、產(chǎn)品細(xì)節(jié)(劑型����、規(guī)格和給藥途徑等)、產(chǎn)品的上市授權(quán)狀態(tài)��、從監(jiān)管機(jī)構(gòu)獲得的在相關(guān)兒科人群開(kāi)發(fā)的建議����、在歐盟是否為孤兒藥��、計(jì)劃提交MAA的日期、研究計(jì)劃簡(jiǎn)介等��。
2.2 疾病概況及產(chǎn)品開(kāi)發(fā)背景
PSP要求分開(kāi)論述兒科疾病的概況及產(chǎn)品開(kāi)發(fā)信息(第1和第2部分)��,而PIP將疾病信息整合到產(chǎn)品開(kāi)發(fā)信息中在Part B部分統(tǒng)一論述��。PSP和PIP都要求針對(duì)擬研究疾病的病理學(xué)生理學(xué)����,疾病的嚴(yán)重程度及疾病在成人與兒童人群中的相似性與差異,目前的診斷��、預(yù)防和治療措施����,兒科人群中的發(fā)病率以及該產(chǎn)品在兒科人群中潛在的治療獲益進(jìn)行討論。
2.3 對(duì)有效性外推研究的概述
從參考人群(如成人或大齡兒童)向目標(biāo)人群(如兒童或低領(lǐng)兒童)的有效性外推����,可以在避免兒童參加不必要的臨床試驗(yàn)的同時(shí)獲得兒科用藥信息。PSP要求在第3部分概述外推計(jì)劃��,而PIP要求在Part D.5部分概述�����。若有外推計(jì)劃則需列出疾病在參考人群與目標(biāo)人群相似性的支持性信息如疾病的發(fā)病機(jī)理,診斷標(biāo)準(zhǔn)�����,病理生理學(xué)��、組織病理學(xué)�����、病理生物學(xué)特征��,暴露量-反應(yīng)關(guān)系及建模和模擬研究等��。
2.4 豁免申請(qǐng)
PSP要求在第4部門(mén)提出豁免申請(qǐng)����,而PIP要求在Part C 部分提出。FDA與EMA對(duì)豁免申請(qǐng)的理由都做出了具體的規(guī)定�����,只有滿(mǎn)足這些規(guī)定才能申請(qǐng)豁免����,同時(shí)要提供支持性信息。
FDA規(guī)定可以申請(qǐng)豁免的標(biāo)準(zhǔn)是:1)必要的研究是不可能或者高度不可行的(如患者數(shù)量極少)��;2)有證據(jù)顯示該產(chǎn)品在全部或者部分兒科患者中是無(wú)效或者不安全的�����;3)該產(chǎn)品與現(xiàn)存的治療方法相比不具有顯著的治療獲益且不可能用于實(shí)質(zhì)數(shù)量的兒童患者��;4)開(kāi)發(fā)適齡的兒科劑型是不可能的�����。EMA規(guī)定可以申請(qǐng)兒科豁免的標(biāo)準(zhǔn)是:1)產(chǎn)品在兒童患者中無(wú)效或不安全�����;2)該藥品打算治療的疾病只發(fā)生在成人中��;3)與現(xiàn)存的兒科患者可獲得的治療相比����,該藥品沒(méi)有顯著的治療獲益?�;砻饪梢允遣糠值?�,即只豁免部分兒科人群的研究。如果該藥品針對(duì)的疾病只發(fā)生在部分兒科患者中或者針對(duì)部分兒科患者開(kāi)發(fā)適宜的劑型是不可能的����。
藥品開(kāi)發(fā)是個(gè)漫長(zhǎng)的過(guò)程,隨著信息的積累�����,豁免可能會(huì)被推翻�����,豁免申請(qǐng)?jiān)赑SP和PIP被批準(zhǔn)時(shí)都只代表監(jiān)管當(dāng)局在當(dāng)時(shí)的決定��,正式的豁免申請(qǐng)?jiān)谧龀錾鲜信鷾?zhǔn)決定時(shí)會(huì)被正式授予��。
2.5延期申請(qǐng)
PSP要求在第5部分提出延期申請(qǐng)�����,而PIP要求在Part E 部分提出��。同豁免一樣延期申請(qǐng)也有特定標(biāo)準(zhǔn)�����,PSP:1)如果在兒科研究完成前該藥品已經(jīng)準(zhǔn)備好用于成人;2)兒科研究應(yīng)該被延期直到獲得足夠的安全和有效性信息����;3)其他合適的理由(如適齡劑型的開(kāi)發(fā)未完成)����。PIP:1)科學(xué)技術(shù)理由;2)與公共健康相關(guān)的理由��;3)開(kāi)始兒科研究之前進(jìn)行成人研究是合適的�����;4)兒科研究花費(fèi)的時(shí)間會(huì)比成人研究長(zhǎng)很多��。在兩個(gè)地區(qū)申請(qǐng)延期的標(biāo)準(zhǔn)是十分相似的����,但經(jīng)濟(jì)因素都不能成為延期申請(qǐng)的理由。同豁免一樣延期申請(qǐng)也只有在批準(zhǔn)上市時(shí)正式授予����。
2.6 列表總結(jié)計(jì)劃的非臨床和臨床研究
PSP要求在第6部分列表總結(jié)所有計(jì)劃的非臨床和臨床研究,而PIP則分散在Part D.3非臨床研究和D.4臨床研究中����。
2.7 適齡劑型開(kāi)發(fā)
若目前的劑型不適用于所有的兒科人群�����,兩個(gè)地區(qū)都要求提供適齡劑型的開(kāi)發(fā)計(jì)劃�����。PSP要求在第7部分概述�����,而PIP分散在Part D.1.3及D.2中����。應(yīng)提供處方����、劑型、規(guī)格��、給藥途徑和輔料信息及開(kāi)發(fā)策略��。PIP還要求所有的質(zhì)量相關(guān)的信息作為獨(dú)立的文件提供。
2.8 非臨床研究
PSP的第8部分與PIP的Part D.1.3和D.3是關(guān)于非臨床研究的內(nèi)容����。在該部分要總結(jié)已有的支持臨床開(kāi)展的非臨床數(shù)據(jù),評(píng)估是否還需要開(kāi)展額外的非臨床研究��。若需要開(kāi)展相關(guān)研究應(yīng)簡(jiǎn)要描述研究計(jì)劃:包括研究類(lèi)型����、物種����,起始劑量,持續(xù)時(shí)間����,給藥途徑及結(jié)果監(jiān)測(cè)方法。
2.9 已有的支持兒科臨床研究設(shè)計(jì)和啟動(dòng)的臨床數(shù)據(jù)
PSP應(yīng)在第9部分簡(jiǎn)要總結(jié)已經(jīng)在成人臨床研究中獲得的安全性��、有效性及藥動(dòng)學(xué)和暴露-反應(yīng)數(shù)據(jù)����,以支持相關(guān)兒童人群的研究。如果有用于篩選兒科劑量的建模和模擬研究計(jì)劃也要包含在該部分中��。PIP與之相對(duì)應(yīng)的信息分散在Part D1.3、D4.1��、D4.4及D5.1部分��。
2.10 臨床研究
2.10.1 兒科藥代動(dòng)力學(xué)/藥效學(xué)研究(PK/PD)
PSP要求在第10.1節(jié)按照第6部分列出的計(jì)劃的臨床研究表的順序討論兒科PK/PD研究��。應(yīng)包含研究類(lèi)型/研究設(shè)計(jì)����、兒科年齡群、擬用的兒科制劑��、劑量范圍����、研究終點(diǎn)及其論證(如PK參數(shù),PD生物標(biāo)志物)����、支持劑量選擇的建模和模擬研究、計(jì)劃的藥物基因組學(xué)分析及樣本量論證��。PIP需要在Part D4.2部分列出研究大綱��,并在D4.5部分提供細(xì)節(jié)����。
2.10.2 臨床有效性和安全性研究
PSP的第10.2節(jié)應(yīng)按照第6部分列出的計(jì)劃臨床研究表的順序概述兒科臨床安全性和有效性研究�����。應(yīng)包括研究類(lèi)型/研究設(shè)計(jì)�����、研究目的��、兒科年齡群�����、入排標(biāo)準(zhǔn)、主要和次要終點(diǎn)�����、終點(diǎn)評(píng)估的時(shí)間點(diǎn)����、統(tǒng)計(jì)學(xué)方法等。PIP需要在Part D4.3部分列出研究大綱��,并在D4.5部分提供細(xì)節(jié),主要概述研究類(lèi)型����、研究設(shè)計(jì)和控制、主要目的�����、研究的兒科年齡群����、最小樣本量、研究用藥信息(劑型�����、劑量����、治療方案及給藥途徑)、主要終點(diǎn)和次要終點(diǎn)及其評(píng)估時(shí)間����、統(tǒng)計(jì)計(jì)劃等。
2.11 研究計(jì)劃時(shí)間表
PSP的第11部分和PIP的Part E.1要求列出所有計(jì)劃進(jìn)行的藥學(xué)��、非臨床和臨床研究時(shí)間點(diǎn)。如方案提交時(shí)間��、研究開(kāi)始時(shí)間��、研究完成時(shí)間��、最終研究報(bào)告提交時(shí)間��。還要提供預(yù)估的上市申請(qǐng)的提交時(shí)間����。同意PSP和PIP不等同于同意完整的臨床研究方案,在臨床研究開(kāi)始前還需要向監(jiān)管當(dāng)局提交完整的試驗(yàn)方案��。
2.12 與其他監(jiān)管機(jī)構(gòu)的兒科研究協(xié)議
PSP要在第12部分提交與其他監(jiān)管機(jī)構(gòu)達(dá)成的兒科研究協(xié)議(如經(jīng)PDCO審評(píng)過(guò)的PIP)��。PIP雖沒(méi)有此要求��,但要求在Part F提供所有支持上述科學(xué)討論相關(guān)的參考文獻(xiàn)����,在此也可以提交其他地區(qū)監(jiān)管機(jī)構(gòu)的建議����,比如FDA的WR��。
三��、歐美兒科藥品研發(fā)計(jì)劃審評(píng)審批流程
3.1 PSP審評(píng)審批流程
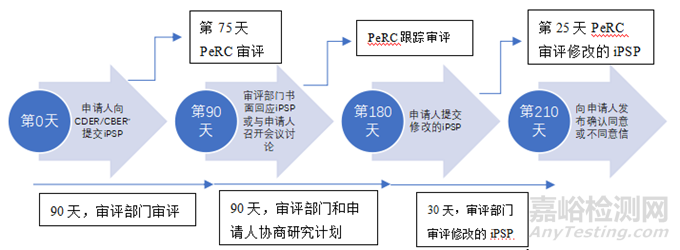
申請(qǐng)人一般應(yīng)該在Ⅱ期臨床結(jié)束(EOP2)會(huì)議召開(kāi)后的60天內(nèi)提交iPSP����;如果沒(méi)有EOP2會(huì)議����,iPSP應(yīng)在Ⅲ期臨床或Ⅱ/Ⅲ期臨床開(kāi)始前召開(kāi)會(huì)議時(shí)提交;若以上都沒(méi)有申請(qǐng)人應(yīng)在預(yù)計(jì)的上市申請(qǐng)?zhí)峤蝗掌诘?10天之前提交iPSP[14]��。
申請(qǐng)人向FDA相關(guān)審評(píng)部門(mén)提交iPSP后�����,審評(píng)部門(mén)有90天的時(shí)間進(jìn)行iPSP的審評(píng)并書(shū)面回應(yīng)或者與申請(qǐng)人召開(kāi)會(huì)議討論�����。其中PeRC對(duì)iPSP的審評(píng)應(yīng)不晚于第75天����。接下來(lái)申請(qǐng)人有第二個(gè)90天去回應(yīng)審評(píng)部門(mén)的要求,期間申請(qǐng)人可以與審評(píng)部門(mén)對(duì)研究計(jì)劃進(jìn)行協(xié)商����,PeRC也會(huì)跟蹤審評(píng)�����,第二個(gè)90天結(jié)束后申請(qǐng)人必須提交修改的iPSP(Agreed iPSP)����。隨后��,F(xiàn)DA有30天時(shí)間審評(píng)修改的iPSP����,PeRC應(yīng)不晚于第25天審評(píng)修改的iPSP并向申請(qǐng)人發(fā)出確認(rèn)同意或者不同意信。審評(píng)審批程序見(jiàn)圖2[17]�����。
*CDER:FDA藥品審評(píng)與研究中心����;CBER:FDA生物制品審評(píng)與研究中心
▲圖3-PSP審評(píng)審批流程及時(shí)間線(xiàn)
3.2 PIP審評(píng)審批流程[18]
申請(qǐng)人應(yīng)在不晚于成人藥代動(dòng)力學(xué)研究完成的日期提交PIP�����,一般為Ⅱ期臨床開(kāi)始之前,不應(yīng)在Ⅲ期臨床試驗(yàn)開(kāi)始后提交��。EMA鼓勵(lì)申請(qǐng)人在人體藥代動(dòng)力學(xué)研究完成之前提交PIP����,尤其是針對(duì)治療嚴(yán)重的或威脅生命的疾病的藥品。
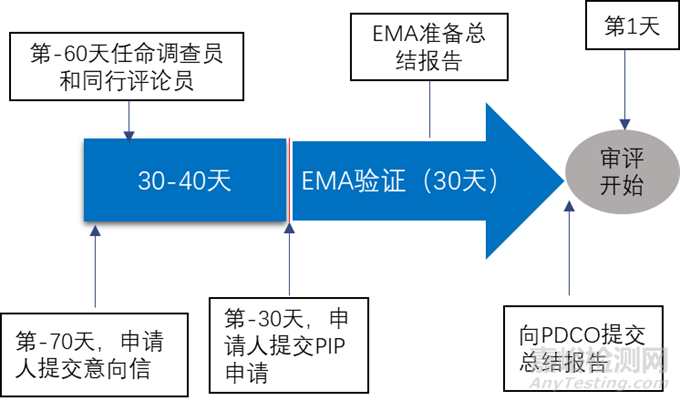
申請(qǐng)人一般需要在正式審評(píng)開(kāi)始前兩個(gè)月提交意向信�����,隨后EMA會(huì)任命一名PDCO (Paediatric Committee) 調(diào)查員與一名PDCO同行評(píng)論員負(fù)責(zé)起草總結(jié)報(bào)告����,申請(qǐng)人在提交前可以申請(qǐng)?zhí)峤磺皶?huì)議。提交申請(qǐng)的時(shí)間約為正式審評(píng)開(kāi)始的前一個(gè)月����,在這一個(gè)月內(nèi)EMA會(huì)對(duì)PIP進(jìn)行驗(yàn)證(validation),主要驗(yàn)證信息的正確性和完整性(行政驗(yàn)證)����,PIP的結(jié)構(gòu)是否正確以及是否有足夠的科學(xué)信息(監(jiān)管與科學(xué)驗(yàn)證)。驗(yàn)證完成后�����,EMA匯總并撰寫(xiě)總結(jié)報(bào)告,并提交給PDCO����,審評(píng)開(kāi)始。
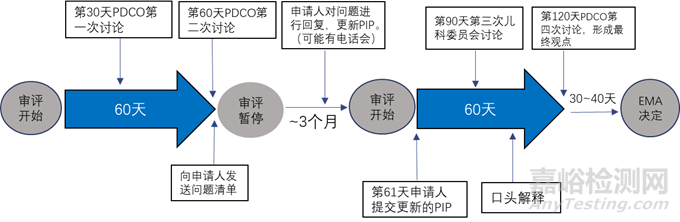
審評(píng)開(kāi)始后的第30天�����,PDCO對(duì)報(bào)告進(jìn)行討論��,并形成總結(jié)報(bào)告�����,總結(jié)報(bào)告中包含PDCO的反饋以及初步觀(guān)點(diǎn)����,該報(bào)告會(huì)發(fā)送其他專(zhuān)家以收集評(píng)論,同時(shí)也會(huì)轉(zhuǎn)發(fā)申請(qǐng)人�����。第60天����,PDCO進(jìn)行第二次討論,若認(rèn)為該P(yáng)IP沒(méi)有問(wèn)題則PDCO審評(píng)程序結(jié)束�����;若認(rèn)為PIP中還有未解決的問(wèn)題則會(huì)向申請(qǐng)人發(fā)送問(wèn)題清單�����,審評(píng)暫停����。審評(píng)暫停期間申請(qǐng)人根據(jù)問(wèn)題清單對(duì)PIP進(jìn)行修改,在此期間可以與PDCO進(jìn)行電話(huà)溝通�����。申請(qǐng)人提交修改的PIP��,審評(píng)開(kāi)始�����,在第90天��,PDCO進(jìn)行第三次討論,主要針對(duì)申請(qǐng)人提交的修訂的PIP��,并形成總結(jié)報(bào)告��。如果還有未解決的問(wèn)題申請(qǐng)人或者PDCO在第三次討論后可要求口頭解釋?zhuān)瓷暾?qǐng)人直接與整個(gè)兒科委員會(huì)對(duì)話(huà)����。PDCO的最終觀(guān)點(diǎn)和最終報(bào)告會(huì)在第120天的第四次討論后發(fā)給申請(qǐng)人。若申請(qǐng)人對(duì)PDCO的最終觀(guān)點(diǎn)有異議�����,可以在30天內(nèi)請(qǐng)求重新評(píng)估��,隨后的10天內(nèi)EMA會(huì)根據(jù)PDCO的最終觀(guān)點(diǎn)和總結(jié)報(bào)告做出決定�����。流程圖見(jiàn)圖4和圖5����。
▲圖4-PIP審評(píng)審批流程(驗(yàn)證階段)
▲圖5-PIP審評(píng)審批流程(評(píng)估階段)
3.3 審評(píng)審批流程對(duì)比
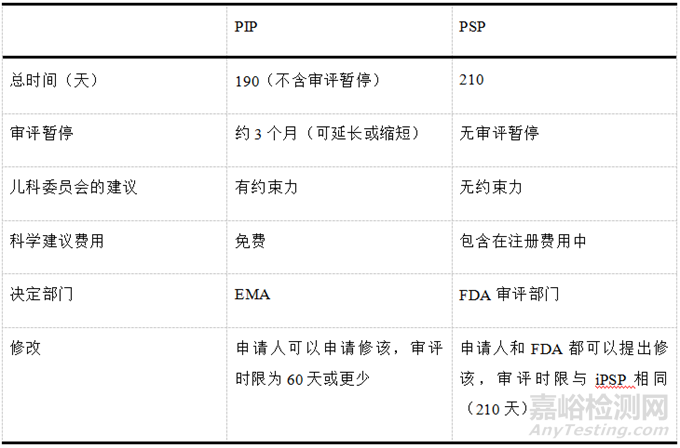
EMA和FDA都鼓勵(lì)申請(qǐng)人盡早提交兒科研發(fā)計(jì)劃,并鼓勵(lì)申請(qǐng)人在提交研究計(jì)劃之前與EMA或FDA溝通�����,以便更好的將兒科研發(fā)計(jì)劃納入整體開(kāi)發(fā)策略。兒科臨床試驗(yàn)逐漸趨向于多區(qū)域多中心臨床試驗(yàn)����,各區(qū)域之間監(jiān)管要求的協(xié)調(diào)統(tǒng)一對(duì)于加快兒科藥品開(kāi)發(fā),避免兒童參與不必要的臨床試驗(yàn)具有重要意義����。分析PSP和PIP的審評(píng)流程�����,存在協(xié)調(diào)統(tǒng)一的可能��。因?yàn)镻IP規(guī)定的提交時(shí)間比PSP的早�����,所以可以先提交PIP�����,在第60天收到PDCO的問(wèn)題清單后��,在隨后PIP的審評(píng)暫停期間����,根據(jù)問(wèn)題清單更新兒科研究計(jì)劃��,向FDA提交iPSP�����,隨后在第90天收到FDA的回饋后����,再更新PIP�����,向PDCO反饋����。雖然PIP的審評(píng)暫停時(shí)限一般為3個(gè)月,但審評(píng)暫停時(shí)限可以根據(jù)具體品種延長(zhǎng)或縮短����,iPSP的審評(píng)流程理論上可以在此審評(píng)暫停時(shí)限內(nèi)開(kāi)始,并在此時(shí)限內(nèi)獲得FDA的反饋建議��,從而規(guī)避部分由于歐美對(duì)兒童用藥臨床試驗(yàn)監(jiān)管要求不同所造成的阻礙�����。PSP與PIP的審評(píng)審批流程對(duì)比見(jiàn)表2。
▲表2-PSP與PIP審評(píng)審批流程對(duì)比
討論
歐美等國(guó)關(guān)于兒童臨床試驗(yàn)的理念已轉(zhuǎn)變?yōu)橥ㄟ^(guò)讓兒童參與臨床試驗(yàn)來(lái)保護(hù)兒童����,制藥企業(yè)已經(jīng)在藥品開(kāi)發(fā)早期就將兒童人群納入整體開(kāi)發(fā)策略。據(jù)報(bào)道EMA已經(jīng)批準(zhǔn)了超過(guò)1000個(gè)PIP申請(qǐng)[12]�����,根據(jù)PERA和BPCA修改說(shuō)明書(shū)中兒童用藥相關(guān)信息的藥品超過(guò)900多種[9]��,極大的促進(jìn)了兒童醫(yī)療保健事業(yè)的進(jìn)步�����。
我國(guó)從2011年發(fā)布《中國(guó)兒童發(fā)展(2011~2020)》以來(lái)開(kāi)始重視兒童用藥的供應(yīng)保障����,近年來(lái)通過(guò)將兒童藥品納入優(yōu)先審評(píng)�����,設(shè)立兒童用藥特殊標(biāo)識(shí)以?xún)?yōu)化審評(píng)資源��,成立兒童用藥專(zhuān)項(xiàng)領(lǐng)導(dǎo)小組和工作小組,完善兒童用藥審評(píng)標(biāo)準(zhǔn)體系�����,開(kāi)展已上市藥品說(shuō)明書(shū)中兒童用藥信息規(guī)范化增補(bǔ)工作等舉措[19]�����,此外國(guó)家藥監(jiān)局近期發(fā)布的《中華人民共和國(guó)藥品管理法實(shí)施條例(修訂草案征求意見(jiàn)稿)》提出12個(gè)月兒童用藥獨(dú)占期��,極大的激勵(lì)了我國(guó)兒童用藥的研發(fā)工作��,2021年全年已有47個(gè)兒童藥品獲批上市[20]�����。
雖然已經(jīng)取得了如此的進(jìn)步��,但應(yīng)意識(shí)到我國(guó)的兒童用藥品種少�����、劑型少����、規(guī)格少�����、特藥少的局面還沒(méi)有完全改善��?���;诖瞬⒔Y(jié)合本文研究?jī)?nèi)容給監(jiān)管當(dāng)局提供以下幾點(diǎn)建議:
1)盡早啟動(dòng)兒童用藥立法工作:從歐美等國(guó)的經(jīng)驗(yàn)不難看出�����,強(qiáng)制性的法律要求以及相關(guān)激勵(lì)和幫扶措施是兒科用藥領(lǐng)域取得進(jìn)步的前提條件�����。我國(guó)應(yīng)盡早啟動(dòng)兒童用藥專(zhuān)項(xiàng)立法工作��,不僅要涵蓋兒童用藥品的研發(fā)和生產(chǎn)����,更要包含流通使用�����、上市后監(jiān)測(cè)、再評(píng)價(jià)等藥品全生命周期的內(nèi)容��。
2)引入兒科研究計(jì)劃: 歐美的兒科法律都引入了專(zhuān)門(mén)的工具(PSP和PIP)以促進(jìn)兒童藥品研發(fā)�����,同時(shí)在其內(nèi)容和審評(píng)審批流程上發(fā)布了相關(guān)指南以指導(dǎo)企業(yè)進(jìn)行兒科研發(fā)�����。我國(guó)可先試點(diǎn)針對(duì)某些特定類(lèi)別的藥品(如超說(shuō)明書(shū)用藥現(xiàn)象嚴(yán)重的藥品)����,制定相關(guān)激勵(lì)措施(如資金支持和技術(shù)指導(dǎo)),在相關(guān)指導(dǎo)原則完善的基礎(chǔ)上強(qiáng)制相關(guān)企業(yè)或研究機(jī)構(gòu)提交兒科研究計(jì)劃����,具體要包括產(chǎn)品開(kāi)發(fā)背景、不同兒科年齡組的安全性和有效性評(píng)估����、豁免和延期申請(qǐng)、外推計(jì)劃�����、已有的支持兒科研究信息、藥學(xué)開(kāi)發(fā)計(jì)劃����、非臨床和臨床開(kāi)發(fā)計(jì)劃及其啟動(dòng)和完成的時(shí)間點(diǎn)。
3)成立兒科藥品審評(píng)委員會(huì):歐美都有專(zhuān)門(mén)的兒科藥品審評(píng)委員會(huì)(PeRC和PDCO)專(zhuān)門(mén)負(fù)責(zé)對(duì)兒科藥品研發(fā)計(jì)劃及其豁免和延期申請(qǐng)進(jìn)行審評(píng)并為審批決定提供建議��,PeRC還要負(fù)責(zé)審評(píng)WR和PPSR��。委員會(huì)成員涵蓋兒科專(zhuān)家��、新生兒專(zhuān)家�����,還有涉及臨床藥理����、統(tǒng)計(jì)��、毒理��、安全性��、化學(xué)��、法律、倫理方面的專(zhuān)家��。我國(guó)在引入兒科研究計(jì)劃的同時(shí)可以成立兒科藥品審評(píng)委員會(huì)負(fù)責(zé)對(duì)兒科研究計(jì)劃的審評(píng)以及制定兒童用藥審評(píng)標(biāo)準(zhǔn)及指導(dǎo)原則��,同時(shí)為申請(qǐng)人提供科學(xué)建議��,促進(jìn)兒童用藥監(jiān)管科學(xué)發(fā)展����。
4)增強(qiáng)國(guó)際合作:歐盟、美國(guó)�����、加拿大�����、日本和澳大利亞等國(guó)的監(jiān)管機(jī)構(gòu)每月會(huì)舉行電話(huà)會(huì)[21]討論收到的關(guān)于兒科研發(fā)計(jì)劃的內(nèi)容��,會(huì)后還會(huì)將會(huì)議上達(dá)成的共識(shí)轉(zhuǎn)告申請(qǐng)人����,以降低監(jiān)管成本,促進(jìn)兒科研發(fā)計(jì)劃的協(xié)調(diào)統(tǒng)一,在避免兒童參與不必要的臨床試驗(yàn)的同時(shí)加快兒童藥品開(kāi)發(fā)����。我國(guó)自2017年加入ICH以來(lái),2018年當(dāng)選ICH管委會(huì)成員��,2021年連任管委會(huì)成員[22]�����,藥品注冊(cè)監(jiān)管制度國(guó)際化明顯����,已能夠在藥品監(jiān)管的國(guó)際舞臺(tái)上發(fā)聲。兒童藥品研發(fā)有趨于多區(qū)域多中心的趨勢(shì)�����,我國(guó)應(yīng)積極參與兒童用藥研發(fā)的國(guó)際合作�����,為兒童藥品研發(fā)的國(guó)際協(xié)調(diào)提供新思路����、新方法、新工具��,同時(shí)鼓勵(lì)跨國(guó)藥企在國(guó)內(nèi)研發(fā)兒童藥品��。
5)加強(qiáng)宣傳教育促進(jìn)理念轉(zhuǎn)變:目前國(guó)內(nèi)大眾對(duì)于兒童參加臨床實(shí)驗(yàn)的接受度不高����,普遍的理念還是通過(guò)避免兒童參加臨床試驗(yàn)來(lái)保護(hù)兒童,而非通過(guò)促進(jìn)兒童參加臨床試驗(yàn)來(lái)保護(hù)兒童����;除了專(zhuān)門(mén)針對(duì)兒童的藥品,制藥公司也未在藥品開(kāi)發(fā)早期就將兒童群體納入藥品開(kāi)發(fā)整體策略中��,而是在成人適應(yīng)癥批準(zhǔn)后才開(kāi)始考慮兒童的適用性�����。監(jiān)管當(dāng)局應(yīng)當(dāng)對(duì)大眾加強(qiáng)宣傳教育����,對(duì)企業(yè)加強(qiáng)溝通交流和培訓(xùn)指導(dǎo),充分利用各學(xué)術(shù)團(tuán)體的影響����,促進(jìn)大眾和制藥企業(yè)理念的轉(zhuǎn)變��。
參考文獻(xiàn)
[1] 史錄文,王曉玲,國(guó)家衛(wèi)生和計(jì)劃生育委員會(huì)兒童用藥專(zhuān)家. 中國(guó)兒童用藥立法研究[M]. 北京:中國(guó)協(xié)和醫(yī)科大學(xué)出版社, 2017.06.
[2] ICH HARMONISED GUIDELINE. ADDENDUM TO ICH E11: CLINICAL INVESTIGATION OF MEDICINAL PRODUCTS IN THE PEDIATRIC POPULATION[EB/OL]. https://database.ich.org/sites/default/files/E11_R1_Addendum.pdf.
[3] 第七次全國(guó)人口普查公報(bào)(第五號(hào))[EB/OL]. http://www.stats.gov.cn/tjsj/tjgb/rkpcgb/qgrkpcgb/202106/t20210628_1818824.html.
[4] 生物醫(yī)藥:政策支持下�����,兒童專(zhuān)用藥步入發(fā)展快速路-研究報(bào)告正文 _ 數(shù)據(jù)中心 _ 東方財(cái)富網(wǎng)[EB/OL]. https://data.eastmoney.com/report/zw_industry.jshtml?infocode=AP202208171577291886.
[5] 許淑紅,張綺,張林琦,等. 探討我國(guó)兒科用藥的發(fā)展現(xiàn)狀及政策層面的思考[J]. 中國(guó)臨床藥理學(xué)雜志, 2020, 36(12): 1760-1767.
[6] 龔傳歡,楊?lèi)?田麗娟. 兒科藥物研發(fā)激勵(lì)政策研究_龔傳歡[J]. 中國(guó)新藥雜志, 2022, 31(11): 1042-1047.
[7] Regulatory Aspects of Paediatric Drug Development. — Scendea[EB/OL]. https://www.scendea.com/regulatory-aspects-of-paediatric-drug-development.
[8] Guidance for Industry Qualifying for Pediatric Exclusivity Under Section 505A of the Federal
Food, Drug, and Cosmetic Act[EB/OL].https://fda.report/media/72029/Qualifying-for-Pediatric-Exclusivity-Under-Section-505A-of-the-Federal-Food--Drug--and-Cosmetic-Act.pdf
[9] Best Pharmaceuticals for Children Act and Pediatric Research Equity Act _ FDA[EB/OL]. https://www.fda.gov/science-research/pediatrics/best-pharmaceuticals-children-act-and-pediatric-research-equity-act.
[10] Federal Court Invalidates FDA Pediatric Rule, Health Law & Policy Institute[EB/OL]. https://www.law.uh.edu/healthlaw/perspectives/Children/021223Federal.html.
[11] 鄭曉瓊. 國(guó)外兒童用藥監(jiān)管現(xiàn)狀[J]. 中國(guó)藥物經(jīng)濟(jì)學(xué), 2011, 28(1): 51-55.
[12] State of Paediatric Medicines in the EU.10 years of the EU Paediatric Regulation[EB/OL]. https://health.ec.europa.eu/system/files/2017-11/2017_childrensmedicines_report_en_0.pdf.
[13] template-scientific-document-part-b-f_en.doc[EB/OL]. https://view.officeapps.live.com/op/view.aspx?src=https%3A%2F%2Fwww.ema.europa.eu%2Fen%2Fdocuments%2Ftemplate-form%2Ftemplate-scientific-document-part-b-f_en.doc&wdOrigin=BROWSELINKEMA.
[14] Pediatric Study Plans_ Content of and Process for Submitting Initial Pediatric Study Plans and Amended Initial Pediatric Study Plans _ FDA[EB/OL]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pediatric-study-plans-content-and-process-submitting-initial-pediatric-study-plans-and-amended.
[15] FDA / EMA Common Commentary on Submitting an initial Pediatric Study Plan (iPSP) and Paediatric Investigation Plan (PIP) for the Prevention and Treatment of COVID-19 [EB/OL]. https:// www.fda.gov/media/138489/download.
[16] Guideline on the format and content of applications for agreement or modification of a paediatric investigation plan and requests for waivers or deferrals and concerning the operation of the compliance check and on criteria for assessing significant studies [EB/OL]. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52014XC0927(01)CELEX_5201.
[17] PMHS Standard Operating Procedure (SOP) for Review of Pediatric Study Plans (PSPs) and Written Requests by the Pediatric Review Committee (PeRC) [EB/OL]. https://www.fda.gov/media/86061/download
[18] PIP assessment procedure[EB/OL]. https://www.ema.europa.eu/en/documents/presentation/presentation-paediatric-investigation-plan-assessment-procedure_en.pdf.
[19] 藥監(jiān)政策速覽(第42期)破解兒童用藥短缺難題[EB/OL]. https://www.cde.org.cn/main/news/viewInfoCommon/6e103cc9209c2c999e1b458a71bb0eb0.
[20] 2021年度藥品審評(píng)報(bào)告[EB/OL]. https://www.cde.org.cn/main/news/viewInfoCommon/f92b7bdf775bbf4c4dc3a762f343cdc8.
[21] International Collaboration _ Pediatric Cluster _ FDA[EB/OL]. https://www.fda.gov/science-research/pediatrics/international-collaboration-pediatric-cluster.
[22] 國(guó)家藥監(jiān)局連任ICH管委會(huì)成員 我國(guó)藥品監(jiān)管加速?lài)?guó)際化進(jìn)階[EB/OL]. https://www.cde.org.cn/main/news/viewInfoCommon/a6f9b5e97e82fb01fe205536d06b2c3f.