為貫徹落實《國務(wù)院關(guān)于改革藥品醫(yī)療器械審評審批制度的意見》(國發(fā)〔2015〕44號),這些年來NMPA連同CDE在簡化藥品審批程序方面出臺了眾多政策和措施�,其中針對原輔包的主要政策就是推行了原輔包關(guān)聯(lián)審評審批制度,主要包括出臺一系列政策文件��、建立原輔包登記平臺、施行原輔包登記管理�、不強求原料藥單獨審評審批��,取消藥用輔料和藥包材行政審批等。
前期����,針對原輔包關(guān)聯(lián)審評審批主題,本人將目光鎖定在國內(nèi)��,推出“原輔包關(guān)聯(lián)審評系列文章”����,計劃從政策變遷及提煉����、核心政策對比與分析、登記資料要求解析��、關(guān)聯(lián)審評審批制度解析����、經(jīng)驗分享與注意事項等方面給大家系統(tǒng)展現(xiàn)原輔包關(guān)聯(lián)審評審批相關(guān)政策理論與實踐經(jīng)驗?,F(xiàn)今����,隨著工作內(nèi)容的擴展,我將視野擴展到世界��,在原系列文章的基礎(chǔ)上進行升級�,推出“原輔包注冊認證系列文章”����,將對包括中國在內(nèi)的�,美國�、歐洲����、日本����、澳大利亞等國家和地區(qū)的相關(guān)法規(guī)制度、認證程序�、以及實踐經(jīng)驗進行分享和展現(xiàn),為大家系統(tǒng)掌握相關(guān)知識提供參考�。
同樣����,由于本人經(jīng)驗有限��,其中不足及有誤之處�,還請大家多多包涵,多多指正����,咱們相互學習�,共同提高,在此致以衷心的感謝�!
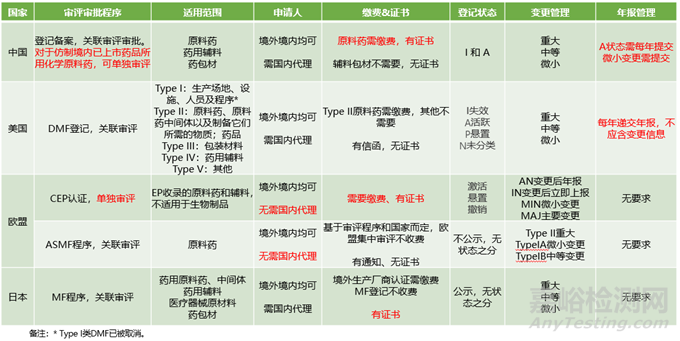
本文我們將通過對一張信息匯總表的解讀,一起了解一下全球最重要的四個國家和地區(qū):中國、美國����、歐洲��、日本����,各自在原輔包注冊認證方面,執(zhí)行哪些制度,各個制度之間有哪些區(qū)別,以便咱們能在全球視野下����,整體了解原輔包注冊認證的基本情況。
話不多說��,讓我們一睹為快�,然后我再對各種制度程序進行一一講解。
一、中國原輔包關(guān)聯(lián)審評審批制度
中國原輔包關(guān)聯(lián)審評的制度��,簡而言之分為兩步:原輔包登記和制劑申報關(guān)聯(lián)審評審批。具體程序分別為:
第一步�,原輔包登記人在CDE原輔包登記平臺上進行登記��,并按要求提交登記資料。CDE審評老師進行形式審查�,通過則在登記平臺上公示,獲得“I”狀態(tài)�;若形式審查有問題����,則按要求補正并再次進行形式審查��,通過后公示,獲得“I”狀態(tài)����。按規(guī)定形式審查應(yīng)在5個工作日完成����,但實際執(zhí)行下來����,由于審核工作量等問題�,有可能會長一些����,一般半個月到1個月之間就能公示����。
第二步�,使用該原輔包的制劑進行申報時,需取得原輔包登記人提供的授權(quán)書,證明制劑研發(fā)過程所使用的原輔包是經(jīng)過相應(yīng)登記人授權(quán)的�,而該授權(quán)書會作為關(guān)聯(lián)審評審批的線索,讓CDE知道在對制劑進行技術(shù)審評時�,需要關(guān)聯(lián)審評哪個登記號的原輔包��。制劑的技術(shù)審評需要200+的時間�,中間還會涉及注冊核查��、注冊檢驗����,補充研究等流程�,耗時較長�。審評審批通過,則制劑獲得批準通知書��,原輔包的登記狀態(tài)變?yōu)?ldquo;A”狀態(tài)��。
此外,針對一些特殊原因無法進行登記的原輔包��,一些仿制境內(nèi)已上市制劑所用原料藥��,以及一些特供某一廠家制劑的原輔包,也可通過與制劑資料一并提供研究資料的方法�,進行申報和審評審批。
基于以上程序�,有幾點需要注意:
1)原輔包登記人:既可以是原輔包生產(chǎn)企業(yè),也可以是原輔包供應(yīng)商��,既可以是國內(nèi)登記人,也可也是國外登記人����。其中國外登記人,需由國內(nèi)代表機構(gòu)或委托國內(nèi)代理機構(gòu)進行登記����。無論是何種身份,作為登記人都需要承擔一定的法律責任�,如保證登記資料真實準確,及時維護登記信息等��。其中針對境外登記人,其與其代理機構(gòu)共同承擔上述法律責任�。
2)登記狀態(tài):以是否通過技術(shù)審評審批為分界線進行區(qū)分�,“I”代表未進行關(guān)聯(lián)審評審批��,或尚未通過關(guān)聯(lián)審評審批��;“A”狀態(tài)代表已經(jīng)通過關(guān)聯(lián)審評審批��。另外還有一些滿足要求的原輔包�,可由CDE自動將其狀態(tài)轉(zhuǎn)換為“A”����,但這類自動轉(zhuǎn)A的原輔包是否能滿足最新的法規(guī)要求����,則還需基于具體關(guān)聯(lián)審評審批而定����。
3)原料藥與藥用輔料�、藥包材的區(qū)別:在中國�,原料藥至今仍需行政審批�,因此針對它�,需要進行關(guān)聯(lián)審評和關(guān)聯(lián)審批兩個步驟�,需要繳費��,審批通過后會有批準通知書;相比之下�,藥用輔料和藥包材的行政審批已被取消,只需關(guān)聯(lián)審評一個步驟��,無需繳費��,也沒有相應(yīng)的批準通知書��。
在中國完成登記,以及關(guān)聯(lián)審評審批的原輔包�,都可以進行變更。變更類型包括基礎(chǔ)信息變更����、重大變更�、中等變更和微小變更��。基礎(chǔ)信息變更屬于非技術(shù)性變更����,主要變更一些非技術(shù)類的信息�,如登記人名稱��、地址等��;而重大、中等和微小變更則為技術(shù)類變更�,針對變更的風險和影響進行界定,可參考《已上市化學藥品藥學變更研究技術(shù)指導(dǎo)原則(試行)》等相關(guān)指導(dǎo)原則進行評估����。
此外�,原輔包登記完成后�,還需要每年第一季度提交上一年的年報����,匯總變更情況����、關(guān)聯(lián)的制劑產(chǎn)品信息等�。此處需要說明一點����,年報僅僅是匯總了變更情況��,不能作為變更的證明性文件。
二�、美國原輔包DMF制度
美國針對原輔包執(zhí)行DMF登記制度�,DMF全稱Drug Master File����,藥品主文件����,是關(guān)于產(chǎn)品CMC信息(化學����、生產(chǎn)�、質(zhì)量控制信息)的一套完整文件資料�,作為一種參閱性資料存檔在FDA中心檔案室(CDR)中����。境內(nèi)外原輔包供應(yīng)商均可進行DMF登記�,其中境外供應(yīng)商需指定美國代理�,而其進行DMF登記的主要目的則是可以在不透露自己保密信息的情況下,被多家制劑企業(yè)引用。
美國DMF分為5類��,分別為:
Type I:生產(chǎn)場地、設(shè)施����、人員及程序(2000年07月已被取消)
Type II:原料藥、原料藥中間體以及制備它們所需的物質(zhì);藥品
Type III:包裝材料
Type IV:藥用輔料
Type V:其他
原輔包供應(yīng)商可基于其產(chǎn)品所屬的類型�,向FDA提交對應(yīng)類型的DMF登記�。FDA在收到資料和申請后����,進行編號備案和行政審評��,通過后就直接給予“A”狀態(tài),一般歷時2~3周����。此階段并不進行技術(shù)審評��,直到與之關(guān)聯(lián)的藥品進行申報時����,才與制劑進行關(guān)聯(lián)審評,審評時間基于制劑的不同注冊類型而定�,以仿制藥為例�,需要大約10個月的時間。除了仿制藥申請引用的Type II類原料藥需要繳費之外,其他DMF登記均無需繳費��,其繳費原因在于,該類型DMF登記會增加一個歷時約60天的完整性審評(即CA審評)階段��,審評通過后進入CA名單。只有進入該名單的原料藥��,才能被制劑仿制藥引用�。
這里有兩點需要注意�,同時也是和中國原輔包關(guān)聯(lián)審評審批程序的兩點區(qū)別:
1)登記狀態(tài)的意義:
DMF制度的狀態(tài)有四種:
A狀態(tài):代表處于維護狀態(tài),可被引用����,但這并不代表已經(jīng)通過技術(shù)審評�;
I狀態(tài):代表失效狀態(tài)�,多由于未及時維護導(dǎo)致�;
P狀態(tài):代表懸停狀態(tài)�,比如行政審評中斷,技術(shù)審評中斷時的狀態(tài)��;
N狀態(tài):代表未分類狀態(tài)��,多在剛剛進入FDA,尚未進行行政審評時的狀態(tài)����。
中國原輔包登記有2種狀態(tài):
A狀態(tài):代表通過技術(shù)審評,已與制劑完成關(guān)聯(lián)審評審批�。
I狀態(tài):代表未通過技術(shù)審評����,比如尚未與制劑關(guān)聯(lián)�,或者處于關(guān)聯(lián)審評過程中。
由此可見����,DMF的A和I狀態(tài),與中國原輔包登記的A和I狀態(tài)�,有著本質(zhì)區(qū)別����。
2)關(guān)聯(lián)審評vs關(guān)聯(lián)審評審批
美國DMF程序只有關(guān)聯(lián)審評�,不存在批準和不批準之說��,不發(fā)證書����,但會發(fā)通知函告知申請人結(jié)果����。
中國原輔包關(guān)聯(lián)審評審批制度,包括審評和審批兩個階段�,其中藥用輔料和藥包材與DMF相同,只需審評�,無需審批,無批準和不批準之說��,但原料藥卻既需要審評,又需要審批�,批準則發(fā)批準通知書,不批準則發(fā)不予批準通知書��。
此外��,在變更和年報方面�,基于變更程度及風險,美國DMF登記變更分為重大��、中等及微小變更��。類似中國原輔包關(guān)聯(lián)審評審批制度,美國DMF也需要提交年報�。
三、歐洲CEP認證制度
歐洲CEP認證,又稱COS認證��,為歐洲藥典適應(yīng)性認證��,適用于被歐洲藥典專論收錄的原料藥和輔料�,也就是說如果沒有被EP專論收錄����,無法進行CEP認證。另外��,目前生物藥不適用于CEP注冊程序。
CEP認證的主管部門為歐洲質(zhì)量監(jiān)督管理局EDQM�。境內(nèi)外原輔包供應(yīng)商都可以進行認證,且沒有歐洲代理的要求��。
CEP認證需要繳費�,采用獨立審評制��,審評通過后頒發(fā)CEP證書,基于認證主體可分為化學CEP�、植物CEP����、TSE產(chǎn)品 CEP����,以及組合CEP認證,各類認證時間不同��,但均能在歐洲范圍內(nèi)通用�,因此常作為原料藥和輔料進入歐洲市場的快速通道�。基于審評和維護情況����,CEP認證目前有激活、懸置和撤銷三種狀態(tài)��。
變更方面��,基于變更的嚴重程度分為四種變更類型����,分別為AN變更后可在年報中通知�,IN變更后立即通知,MIN微小變更和MAJ重要變更��。
年報方面,CEP認證不強求進行年報����,但有一次五年再注冊的要求�,再注冊后可永久有效����。
四����、歐洲原料藥ASMF登記制度
除了CEP認證之外,歐洲還有一個僅針對歐洲原料藥的ASMF登記程序��,該程序主管部門為歐洲藥品管理局EMA��,境內(nèi)外供應(yīng)商均可以申請����,無代理要求��。該程序類似于美國DMF登記程序,相同點是都采用關(guān)聯(lián)審評的方式在制劑申報時一并審評����,具體時限基于歐洲不同的申報流程而定����;不同點是該登記程序僅針對歐洲上市的原料藥�,不適用于藥用輔料和生物制劑,而且不公示�,不區(qū)分狀態(tài),也不發(fā)證書��,只發(fā)通知函告知登記情況�。
變更方面,分為兩大類三小類����,Type IA為微小變更,Type IB為中等變更����,Type II為重大變更。此外�,該程序沒有年報要求。
五��、日本原輔包MF登記制度
在日本��,原輔包管理采用MF登記程序����,即注冊主文件登記程序,該程序的適用范圍包括藥用原料藥��,還適用于中間體��、藥用輔料��、醫(yī)療器械原材料和藥包材等��。其主管部門為厚生勞動省下屬的PMDA�,日本厚生勞動省相當于中國的的NMPA,PMDA相當于中國的CDE����。
日本境內(nèi)外供應(yīng)商都可以進行登記,但是境外供應(yīng)商必須指定日本代理�,若生產(chǎn)廠在國外,還需要進行國外生產(chǎn)廠確認程序AFM��,并獲得認定證書和廠商編碼BN����。MF登記程序本身不收費,但AFM確認程序則需要繳費��,并AFM證書需5年更新一次。
其實����,日本MF登記程序并非法定必須的程序,而是一種自愿行為��。MF登記程序也不存在批準或不批準�,MF登記證也不能代表銷售證明。遞交申請后��,PMDA只進行形式審查�,通過后發(fā)放原料藥登記證和登記申請書副本,并在PMDA官網(wǎng)上定期公示登記信息��,目前更新頻率為每月兩次��,分別在月中和月底��。
日本MF登記程序��,采用關(guān)聯(lián)審評制�,只有在制劑廠商引用該原料藥時才會進行技術(shù)審評并實施GMP檢查,審評時限基于藥物制劑的申報類型而定��。
變更和年報方面����,MF登記程序分為重大�、中等��、輕微三類變更����,無年報要求�。