在2022年11月���,由RDPAC 的藥學團隊對中美歐藥學技術指導原則和指南進行了調(diào)研與對比�����,總結(jié)分析了中美歐指南的標準差異及實施情況差異���,采用了主題詞及分類方式���、深度對比���、報告撰寫和定稿流程�����,最后呈現(xiàn)研究報告�����,給到藥審中心參考��。
隨著全球同步研發(fā)理念的推進�����,創(chuàng)新藥的全球申報也逐漸變得同步��,符合我國新注冊管理 辦法全球新注冊分類的申報也逐漸增多�����。同時�����,工藝驗證是產(chǎn)品工藝生命周期中的重要環(huán)節(jié)��, 充分全面的工藝驗證對保障產(chǎn)品安全有效質(zhì)量可控必不可少�����。我國藥品相關管理法規(guī)��、GMP 中產(chǎn)品生產(chǎn)工藝驗證的要求�����,與 ICH 主要國家監(jiān)管趨同�����。
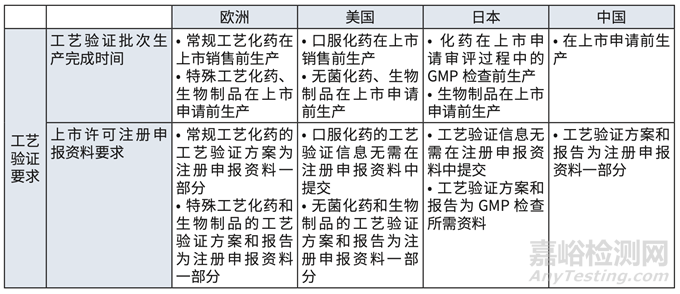
但是對比歐美日等國家及我國對工藝驗證批次生產(chǎn)完成時間�����、上市許可申報資料要求等可 見,我國要求工藝驗證批次的生產(chǎn)發(fā)生在上市申請前��,工藝驗證方案及報告�����,兩者均作為注冊 申報資料的一部分�����。工藝驗證完成時間點���,何時遞交及遞交內(nèi)容等,影響了全球同步研發(fā)��。
為保證境外生產(chǎn)藥品在境內(nèi)外的同步亦或首先申報和獲批�����,建議在相關指南中���,或在問與 答(Q&A)等�����,或在相關培訓中���,對在上市申請中�����,適當減免對于部分區(qū)域性文件(例如驗證報告���, 批記錄等)的要求,在批準后隨年報遞交或在檢查時重點確認��?��;蛘甙l(fā)布工藝驗證相關管理文 件��,統(tǒng)一不同法規(guī)中對工藝驗證的要求��,應用 ICHQ8-11 主要概念和引入“連續(xù)工藝驗證確認” 的概念���,并相應的調(diào)整遞交文件的要求。
中國及歐美現(xiàn)行法規(guī)文件對比:
對比歐美日等國家及我國對工藝驗證批次生產(chǎn)完成時間�����、上市許可申報資料要求等,我國要求工藝驗證批次的生產(chǎn)發(fā)生在上市申請前��,工藝驗證方案及報告��,兩者均作為注冊申報資料的一部分�����。
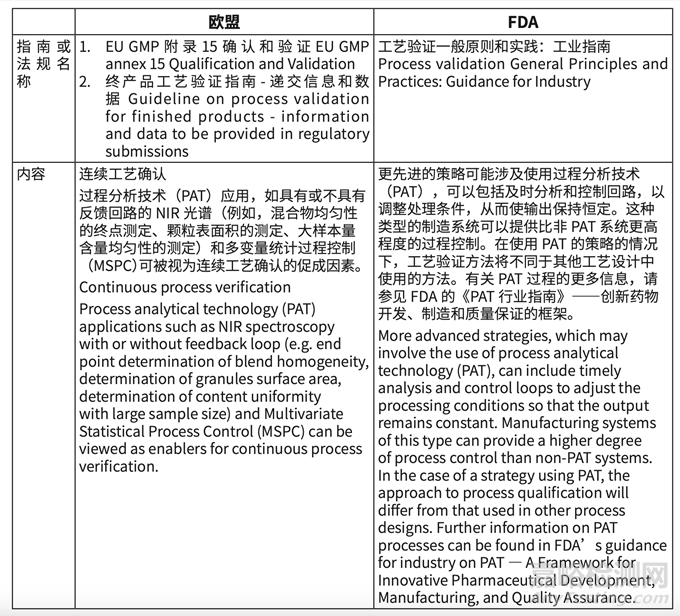
如表格 3 的指南比對的結(jié)果��,中國缺少 EMA 的成品工藝驗證指南 - 監(jiān)管文件中提供的信 息和數(shù)據(jù)(Guideline on process validation for finished products-information and data to be provided in regulatory submissions)和生產(chǎn)生物技術活性物質(zhì)的工藝驗證和監(jiān)管提交文 件 中 提 供 數(shù) 據(jù) 的 指 南(Process validation for the manufacture of biotechnology-derived active substances and data to be provided in the regulatory)�����,所以驗證信息的遞交期待和原液的驗證具體要求不清晰��。
根據(jù)《生物制品注冊受理審查指南》法規(guī)���,產(chǎn)品上市申報時,被要求提供如工藝驗證方案和報告���,批記錄 / 檢驗記錄�����,分析方法驗證報告�����,穩(wěn)定性圖譜等��。但參考境外的要求�����,對某些產(chǎn)品上述部分研究并未要求開展且對審評的參考價值有限��,尤其工藝驗證報告��。而且根據(jù) ICH 的協(xié)調(diào)結(jié)果���,目前提供的 CTD 資料���,以及在模塊一中遞交的“生產(chǎn)工藝信息表”及“藥 品注冊標準”,已基本包含國際公認的產(chǎn)品上市審評所需信息�����。
比如,歐洲指南指出小規(guī)模研究可用于工藝驗證的不同研究���,并說明可以考慮小規(guī)模研究的情況(例如瀝濾物和下游工藝研究等等)�����,同時��,明確提出多場地生產(chǎn)(Multifacility production)必須注意的原則與要求���。
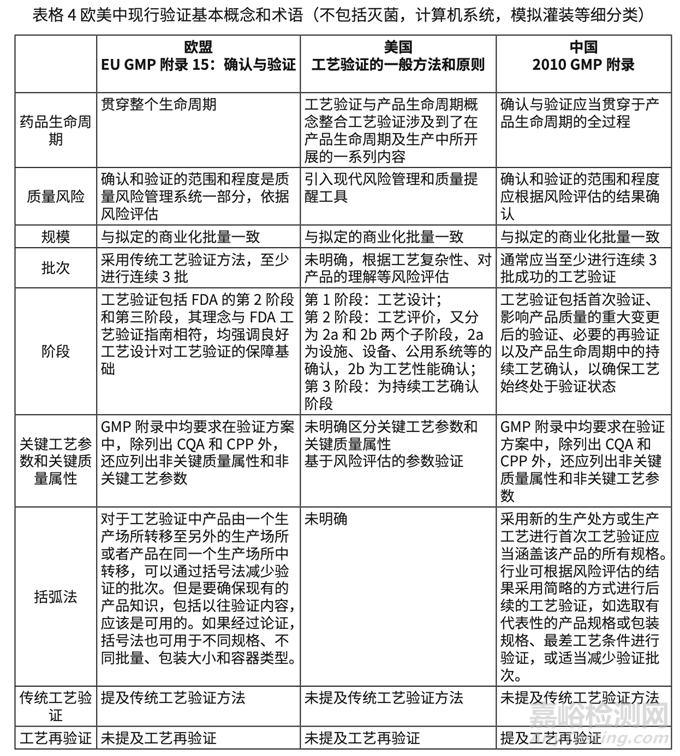
1. 基本概念和術語比對:
1)美國術語不同,將 Process Design 也包括在 Process validation 下���,并在內(nèi)容和理念上強調(diào)了各個階段的相互關系���。歐洲和中國基本理念相同,雖然都有研發(fā)設計理念(ICH Q8)��, 但是都沒放在確認和驗證概念下��。在實踐層面��,可能歐美在研發(fā)階段工作更全面���,而我國對工藝驗證(FDA 第二階段)相對監(jiān)管期待更高�����。
在藥品全生命周期的理念及我國倡導創(chuàng)新的需求下��,F(xiàn)DA 對驗證三階段的明確描述�����,更加值得借鑒�����。
2)歐洲明確提出工藝驗證有傳統(tǒng)工藝驗證���,連續(xù)工藝確認(Continuous process verification)和混合方式,美國沒有明確提出概念以區(qū)分不同的驗證方式(注美國術語 continuous process verification 與歐洲的內(nèi)涵不同���,是第三階段���,詳見下表,但是文字描述 上也有提出了工藝驗證(FDA 術語 PPQ)的不同方式���,比如 PAT 的驗證不同�����。
鑒于連續(xù)工藝確認和持續(xù)深入的研發(fā)(Q8)以及連續(xù)生產(chǎn)(Q13)等存在內(nèi)在聯(lián)系���,歐洲方式更加值得借鑒��。
3)歐美都明確提出了驗證和 ICH Q8, 9, 10, 11 概念的一致�����。
2. 其他
通過比對發(fā)現(xiàn)我國的 GMP 附錄確認和驗證與 EU GMP Annex 15 高度協(xié)調(diào)但是有一些基本原則(principle 和 general 部分)沒有明確表述�����。比如對于驗證批次的問題���,中歐都是基于風險至少三批,但是歐洲在工藝驗證的一般原則部分明確提出可以使用括號法��,并說明可以考慮 括號法的情況�����。具體各個部分的詳細內(nèi)容���,見表格 4��。
中國現(xiàn)存在的問題:
1. 盡管 NMPA 已公告推薦適用 ICH Q8~11 ���,但其概念和原則尚未在驗證領域中得以良好實施。比如:
1)Q8, 11 基于質(zhì)量源于設計(QbD)�����,研發(fā)階段工作的監(jiān)管期待需明確���,要求需強化�����。
2)Q10 基于全生命周期的理念���,明確研發(fā)對驗證的影響以及兩者之間的銜接。
3)Q9 明確基于風險的驗證工作��。
2. 潛在由于以上理念的實施和監(jiān)管期待不明確,我國對于驗證的要求傾向于一刀切�����,而無法基于產(chǎn)品和工藝的風險來進行���,可能無法具有監(jiān)管的靈活性���。比如:
1)驗證都要在注冊遞交前完成,而歐美低風險產(chǎn)品和工藝可在上市前完成�����。(詳見表格 5)

2)批次數(shù)量基于風險可以使用括號法等(詳見表格 6)
3. 我國對傳統(tǒng)工藝驗證的要求較為充分�����,但是替代方式(連續(xù)工藝確認��,EU 概念 continuous process verification)沒有描述��。傳統(tǒng)工藝驗證的替代方式��,主要應用于連續(xù)生產(chǎn)包括相關的 PAT(Process Analytical Technology)和 RTRT(Real Time Release Test)��。缺乏相關指南,對我國未來新技術的應用可能產(chǎn)生一定影響��。國外要求描述(詳見表格 7)��。
表格 7 連續(xù)工藝確認的概念和描述
• 制定新指南可以給工業(yè)界和監(jiān)管機構帶來的益處
第一��、全面深入落實 ICH 的基本概念�����,強化產(chǎn)品開發(fā)階段���,強化 QbD 理念,將基于風險和全生命周期的理念真正貫穿于產(chǎn)品開發(fā)到驗證��,到驗證狀態(tài)的維護(FDA 的階段一�����、二�����、三)��。 從而從根本上保證了產(chǎn)品質(zhì)量,提高了行業(yè)的質(zhì)量管理水平���。
第二�����、在此基礎上��,監(jiān)管基于風險而具有一定靈活性成為了可能�����。在靈活的監(jiān)管下�����,產(chǎn)業(yè)發(fā)展得到了促進�����,產(chǎn)品更快上市�����,利于患者及時快速穩(wěn)定獲得高質(zhì)量的產(chǎn)品��。依據(jù)產(chǎn)品類別和 生產(chǎn)工藝的風險情況���,對工藝驗證的完成時間點和遞交信息���,進行差異化管理,也成為了可能���, 更加快速的讓產(chǎn)品惠及患者也成為了可能。
第三�����、國際協(xié)調(diào)有利于產(chǎn)品的同步研發(fā)���,同步在中國上市���。同時也有利于中國行業(yè)進一步 加入國際供應鏈,參與國際競爭�����。
• 相關的國內(nèi)監(jiān)管法規(guī) :
《注冊管理辦法》第三十四條申請人在完成支持藥品上市注冊的藥學��、藥理毒理學和藥物 臨床試驗等研究,確定質(zhì)量標準�����,完成商業(yè)規(guī)模生產(chǎn)工藝驗證��,并做好接受藥品注冊核查檢驗 的準備后���,提出藥品上市許可申請�����,按照申報資料要求提交相關研究資料���。
建議:
如果我國的工藝驗證要求和監(jiān)管期待與國際普遍實踐存在差異,比如�����,全球工藝驗證尚未完成��,歐美已接受遞交而我國遞交要等到工藝驗證之后�����,那么這將潛在影響創(chuàng)新藥的全球同步研發(fā),并造成全球新的創(chuàng)新藥在我國同步上市的可能性降低�����,影響全球新的創(chuàng)新藥在中國上市���,惠及中國患者���。所以建議出臺相關文件細化以下要求:
1. 檢查部門針對 ICH Q8、9�����、10���、11 原則在工藝驗證領域的落實應用,強化的研發(fā)方式 (Enhanced approach)驗證的替代方法等�����,給與明確指導�����。
2. 細化監(jiān)管要求。建議內(nèi)容包括�����,基于產(chǎn)品類別和工藝判定風險的基本原則;依據(jù)風險 確定工藝驗證完成時間和遞交信息的具體要求;括號法的應用等��。
3. 為保證及時將藥品惠及中國患者���,建議在產(chǎn)品上市申報及某些變更申請時�����,免除或優(yōu)化工藝驗證相關資料的申報要求��?��?筛鶕?jù)需要在年報或藥品現(xiàn)場檢查時提供?��;蛘咴谏鲜猩陥髸r要 求遞交工藝驗證方案���,批準后按年報或按承諾時間遞交報告。