在2022年11月��,由RDPAC 的藥學(xué)團(tuán)隊(duì)對(duì)中美歐藥學(xué)技術(shù)指導(dǎo)原則和指南進(jìn)行了調(diào)研與對(duì)比,總結(jié)分析了中美歐指南的標(biāo)準(zhǔn)差異及實(shí)施情況差異��,采用了主題詞及分類方式、深度對(duì)比���、報(bào)告撰寫和定稿流程��,最后呈現(xiàn)研究報(bào)告�����,給到藥審中心參考。
參考?xì)W美已有的指導(dǎo)原則��,課題組對(duì)我國(guó)目前尚未建立的指導(dǎo)原則或即使我國(guó)有相關(guān)指南���,但要求存在差異的情況�����,進(jìn)行了梳理。其中考慮我國(guó)監(jiān)管體系中法律法規(guī)��、規(guī)章制度等情況��, 如下共計(jì) 20 個(gè)問題屬于不能通過藥審中心發(fā)布的技術(shù)指導(dǎo)原則能解決的問題��,需要單獨(dú)成立 相關(guān)課題進(jìn)行研究�����。為了便于日后相關(guān)管理部門后續(xù)制修訂法規(guī)時(shí)進(jìn)行參考��,課題組匯總這些問題的描述�����、境內(nèi)外法規(guī)指南的對(duì)比��,以及初步擬定的監(jiān)管建議,以供參考���。問題如下:
1. 藥品注冊(cè)檢驗(yàn)相關(guān)問題
2. 工藝驗(yàn)證研究相關(guān)問題
3. 我國(guó)區(qū)域藥學(xué)資料要求存在的問題
4. 我國(guó)藥品質(zhì)量標(biāo)準(zhǔn)建立存在的問題
5. 我國(guó)對(duì)殘留溶劑及元素雜質(zhì)等與 ICH 協(xié)調(diào)的問題 6. 我國(guó)對(duì)雜質(zhì)與 ICH 協(xié)調(diào)的問題
7. 我國(guó)病毒安全性評(píng)價(jià)與 ICH 協(xié)調(diào)的問題
8. 在 ICH 框架下��,我國(guó)與境外其他藥典協(xié)調(diào)的問題 9. 我國(guó)動(dòng)物細(xì)胞制備及質(zhì)量控制與 ICH 協(xié)調(diào)的問題 10. 我國(guó)穩(wěn)定性研究與 ICH 協(xié)調(diào)的問題
11. 實(shí)時(shí)檢驗(yàn)放行和參數(shù)放行相關(guān)問題
12. ICH Q8-11 的落地實(shí)施
13. ICH Q12 的落地實(shí)施
14. 為了維護(hù)藥品持續(xù)供應(yīng)的監(jiān)管特別程序問題
15. GMP 與交叉污染控制等問題
16. 我國(guó)注射用水工藝及標(biāo)準(zhǔn)要求問題
17. 細(xì)化批準(zhǔn)前生產(chǎn)的批次上市要求問題
18. 推進(jìn)原輔包與制劑的關(guān)聯(lián)審評(píng)審批問題
19. 細(xì)化基于風(fēng)險(xiǎn)的境外核查
20. 減免境外生產(chǎn)產(chǎn)品變更申請(qǐng)證明性文件要求
注冊(cè)檢驗(yàn)相關(guān)法規(guī)文件
1.檢驗(yàn)啟動(dòng)程序
注冊(cè)檢驗(yàn)是我國(guó)藥品注冊(cè)申請(qǐng)審評(píng)審批的重要監(jiān)管手段���,但是當(dāng)前缺乏相關(guān)細(xì)化要求���,行 業(yè)很難在變更之前對(duì)該變更是否會(huì)觸發(fā)注冊(cè)檢驗(yàn)做出合理完善的計(jì)劃��,受注冊(cè)檢驗(yàn)的樣品、對(duì) 照品和試劑等物料的訂購進(jìn)口清關(guān)等準(zhǔn)備工作的影響���,延遲了這類申請(qǐng)的批準(zhǔn)���,增加了產(chǎn)品的 斷貨風(fēng)險(xiǎn),危及患者用藥��。
為此�����,我國(guó)推薦依據(jù)上市產(chǎn)品和其變更的技術(shù)風(fēng)險(xiǎn)�����,從科學(xué)角度�����,討論并制定藥品注冊(cè)檢 驗(yàn)啟動(dòng)程序。該文件將對(duì)注冊(cè)檢驗(yàn)啟動(dòng)觸發(fā)標(biāo)準(zhǔn)及具體檢驗(yàn)范圍等方面給予切實(shí)指導(dǎo),進(jìn)一步 助力注冊(cè)檢驗(yàn)啟動(dòng)工作的科學(xué)性和透明度�����。
原因分析
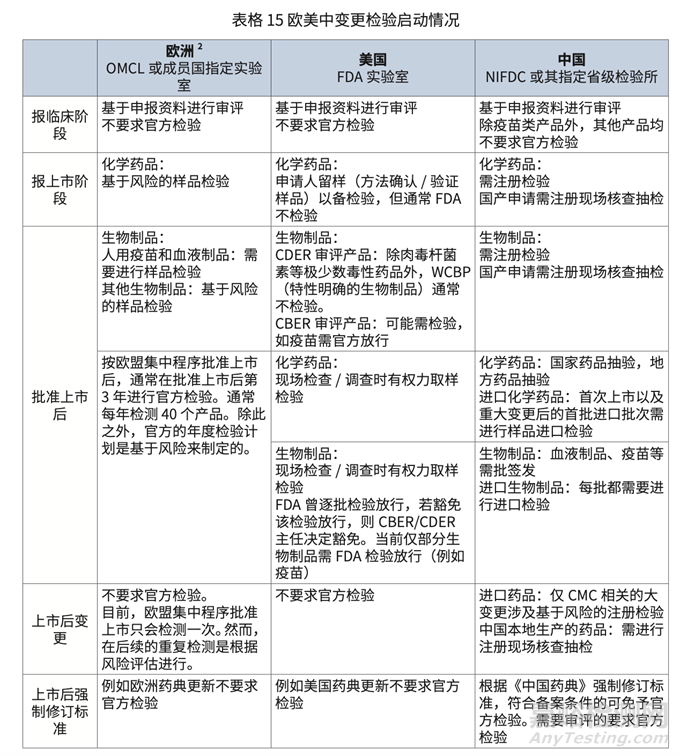
• 歐美現(xiàn)行指南情況梳理:本文依據(jù)藥品上市不同階段對(duì)歐美中的變更檢驗(yàn)情況進(jìn)行梳 理��,詳細(xì)內(nèi)容請(qǐng)見表格 15。
中國(guó)現(xiàn)存在的問題:
中國(guó)目前發(fā)布的《藥品注冊(cè)核查檢驗(yàn)啟動(dòng)工作程序》(2021 年第 54 號(hào)文) 規(guī)定了注冊(cè)檢驗(yàn)的啟動(dòng)流程��,但沒有明確基于風(fēng)險(xiǎn)的原則。目前我國(guó)尚無相關(guān)文件來指導(dǎo)行業(yè) 或監(jiān)管機(jī)構(gòu)��,如何基于風(fēng)險(xiǎn)啟動(dòng)注冊(cè)檢驗(yàn)。由于上市后變更的批準(zhǔn)時(shí)間對(duì)后續(xù)產(chǎn)品供應(yīng)的影響 較大,故這個(gè)領(lǐng)域哪些變更會(huì)引發(fā)注冊(cè)檢驗(yàn)的啟動(dòng)��,問題比較突出�����,具體挑戰(zhàn)如下:
1. 注冊(cè)檢驗(yàn)的目的不明確:作為一種監(jiān)管手段�����,注冊(cè)檢驗(yàn)的目的是什么���,它可以幫助控 制哪些風(fēng)險(xiǎn)�����,如何控制這類風(fēng)險(xiǎn)��。這些管理目的尚未明確��。
《藥品注冊(cè)管理辦法》強(qiáng)調(diào)應(yīng)基于風(fēng)險(xiǎn)啟動(dòng)注冊(cè)檢驗(yàn),但判斷風(fēng)險(xiǎn)高低的標(biāo)準(zhǔn)是尚無統(tǒng)一 的行業(yè)和監(jiān)管的認(rèn)識(shí)��。表現(xiàn)在審評(píng)強(qiáng)烈的依賴注冊(cè)檢驗(yàn)��。以“增加或變更一個(gè)新(非“A”登 記狀態(tài))輔料供應(yīng)商”為例�����,這類變更的審評(píng)是否一定需要注冊(cè)檢驗(yàn)�����,使用了寶貴的監(jiān)管資源 進(jìn)行了注冊(cè)檢驗(yàn),對(duì)審評(píng)的價(jià)值和貢獻(xiàn)都有哪些�����,都需深思���。
2. 注冊(cè)檢驗(yàn)的范圍要求不明確�����。例如�����,
1)對(duì)“注冊(cè)檢驗(yàn)”�����、“樣品檢驗(yàn)”��、“標(biāo)準(zhǔn)復(fù)核”和“單項(xiàng)或部分項(xiàng)目復(fù)核”的定 義和檢驗(yàn)通知單中的描述不明確�����,或在實(shí)際使用中措辭欠嚴(yán)謹(jǐn);
2)由于原液 / 原料藥的變更導(dǎo)致藥檢時(shí)��,建議主要以變更所涉及的原液 / 原料藥的檢 驗(yàn)為主��,并明確必須觸發(fā)制劑藥檢的前提條件;
總之�����,這使得行業(yè)很難在變更之前對(duì)該申請(qǐng)觸發(fā)的注冊(cè)檢驗(yàn)做出合理完善的計(jì)劃(如注冊(cè) 檢驗(yàn)的樣品��、對(duì)照品和試劑等物料的協(xié)調(diào)準(zhǔn)備工作等)���,從而導(dǎo)致整個(gè)審評(píng)審批時(shí)限的延長(zhǎng)��, 增加產(chǎn)品的斷貨風(fēng)險(xiǎn)��。
帶來的益處:
對(duì)現(xiàn)有的上市后變更研究指導(dǎo)原則很好的補(bǔ)充,可加 深行業(yè)對(duì)上市后變更申請(qǐng)要求的理解��,提高行業(yè)共識(shí)�����。能更好地指導(dǎo)行業(yè)做好變更申請(qǐng)的準(zhǔn)備 工作,也充分體現(xiàn)了公開透明的審評(píng)理念��。此外��,也能在一定程度上減輕藥檢機(jī)構(gòu)的工作負(fù)荷 和藥審中心審評(píng)時(shí)限的壓力��。合理地縮短整個(gè)申請(qǐng)審評(píng)的辦結(jié)周期���,降低由于審評(píng)審批周期延 長(zhǎng)導(dǎo)致的產(chǎn)品斷供(尤其是罕見疾病和特殊產(chǎn)品)風(fēng)險(xiǎn)��。從根本上推動(dòng)行業(yè)工藝質(zhì)量?jī)?yōu)化的進(jìn)程��, 讓更好的產(chǎn)品更快速地惠及我國(guó)患者�����。
目前我國(guó)相關(guān)監(jiān)管法規(guī)及指南文件:
- 《藥品注冊(cè)檢驗(yàn)工作程序和技術(shù)要求規(guī)范(試行)》
- 《藥品注冊(cè)核查檢驗(yàn)啟動(dòng)工作程序(試行)》
- 《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》
- 《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》
建議:
考慮到中國(guó)的 GMP 已趨于完善�����,加入 ICH 并也實(shí)施了多個(gè) ICH Q 和 M 指導(dǎo)原則���,中國(guó)現(xiàn) 在的制藥行業(yè)已具備了更多的經(jīng)驗(yàn)和更完善的質(zhì)量管控。建議參考?xì)W美的對(duì)注冊(cè)檢驗(yàn)的管理經(jīng) 驗(yàn)��,制定基于風(fēng)險(xiǎn)的藥品注冊(cè)檢驗(yàn)啟動(dòng)程序。
在上市申請(qǐng)時(shí)���,建議考慮基于風(fēng)險(xiǎn)的注冊(cè)檢驗(yàn)。對(duì)于上市后變更���,考慮到行業(yè)已在上市時(shí) 對(duì)產(chǎn)品進(jìn)行了檢驗(yàn)并有市場(chǎng)抽檢等其他監(jiān)管方式��,建議可以參考行業(yè)的檢驗(yàn)資料作為審評(píng)依據(jù)���, 不要求注冊(cè)檢驗(yàn)���。如需檢驗(yàn)��,也應(yīng)考慮結(jié)合現(xiàn)有的上市后變更研究指導(dǎo)原則,依據(jù)變更的風(fēng)險(xiǎn)�����, 從科學(xué)性角度制定藥品注冊(cè)檢驗(yàn)啟動(dòng)程序���。
建議基于風(fēng)險(xiǎn)考量注冊(cè)檢驗(yàn)的必要性,并明確在何種前提下進(jìn)行哪種等級(jí)的注冊(cè)檢驗(yàn)(“樣品檢驗(yàn)”���、“標(biāo)準(zhǔn)復(fù)核”或“單項(xiàng)或部分項(xiàng)目復(fù)核”)��,并給出推薦的檢驗(yàn)樣品(制劑和 / 或原料藥 / 原液)批數(shù)要求(例如一批還是三批)���,以上信息均在檢驗(yàn)通知單上注明�����。在指南起草過程當(dāng)中�����,邀請(qǐng)工業(yè)界參與討論的機(jī)會(huì)。
由于二次檢驗(yàn)會(huì)嚴(yán)重拖延影響惠及病人的時(shí)間,對(duì)二次檢驗(yàn)也需有合適的風(fēng)險(xiǎn)評(píng)級(jí)���,建議 只有在可能影響產(chǎn)品安全�����、有效和質(zhì)量可控性的情況下才應(yīng)觸發(fā)二次藥檢��。
罕見病用藥(包括細(xì)胞/基因治療)的檢驗(yàn)啟動(dòng)
總結(jié):
現(xiàn)缺乏對(duì)于罕見病用藥(包括細(xì)胞 / 基因治療)的檢驗(yàn)啟動(dòng)條件指南�����。然而通常這類藥品 難以符合滿足目前通用的注冊(cè)檢驗(yàn)要求�����,因此影響延誤了此類產(chǎn)品進(jìn)入中國(guó)并惠及中國(guó)患者的 可能性��。推薦加急制定���。
原因分析:
歐美現(xiàn)行指南
對(duì)于罕見病用藥(包括細(xì)胞 / 基因治療):因?yàn)閲?guó)外的注冊(cè)檢驗(yàn)通常是基于產(chǎn)品特性和風(fēng) 險(xiǎn)而確定的���,通常也不是強(qiáng)制要求��。罕見病及細(xì)胞基因治療用產(chǎn)品,本身市場(chǎng)小產(chǎn)量低���,尚需 鼓勵(lì)��,不能滿足基于注冊(cè)檢驗(yàn)的審評(píng)需要���。
中國(guó)現(xiàn)存問題:
目前中國(guó)對(duì)于罕見病用藥(包括細(xì)胞 / 基因治療)都遵照現(xiàn)行其他藥品的檢驗(yàn)要求執(zhí)行���, 即:要求申請(qǐng)人提供:三批原液 / 原料藥以及三批制劑�����,每批樣品均需是商業(yè)生產(chǎn)規(guī)模的批準(zhǔn)次, 且這 6 批樣品均需要提供三倍檢驗(yàn)用量���,滿足至少 6 個(gè)月(僅做樣品檢驗(yàn))/9 個(gè)月(做樣品 檢驗(yàn)和方法復(fù)核)的剩余效期��。不僅如此��,樣品進(jìn)口通關(guān)時(shí)同時(shí)需要滿足 6 個(gè)月(產(chǎn)品全效期 不足 12 個(gè)月時(shí))/12 個(gè)月(產(chǎn)品全效期多于 12 個(gè)月時(shí))的剩余效期。
然而���,罕見病用藥(包括細(xì)胞 / 基因治療)���,由于年產(chǎn)量小�����,批次少��,往往難以達(dá)到。細(xì) 胞 / 基因治療制品也因?yàn)槿缦绿攸c(diǎn)��,很難滿足檢驗(yàn)要求:
1) 批量問題:批量小,生產(chǎn)及放行檢測(cè)周期長(zhǎng)�����,全年生產(chǎn)量小(對(duì)于一些需要自體細(xì)胞 的產(chǎn)品���,由于自體細(xì)胞的獲得量有限��,批量和產(chǎn)量會(huì)更加有限)���,有一些產(chǎn)品單批批 量在完成行企業(yè)自己的內(nèi)部放行檢驗(yàn)后可能都無法滿足一次官方檢驗(yàn)的量;
2) 效期問題:產(chǎn)品效期普遍較短(一些藥物從診斷試劑檢測(cè)結(jié)果或獲取自體細(xì)胞之后到 給藥的間隔可能是以周為單位來計(jì)算)。
3) 方法問題:檢測(cè)方法往往比較復(fù)雜,方法成功轉(zhuǎn)移到官方檢測(cè)實(shí)驗(yàn)室所需的時(shí)常也往 往會(huì)更久��。
4) 成本問題:由于這類藥物往往價(jià)值較高��,巨大的官方檢驗(yàn)樣品量(尤其一些基于藥典 要求的普適性檢查:如外觀檢查���,異常毒性���,裝量,無菌等)意味著巨大的成本投入 并且��。這些樣品本來是可以惠及患者�����。
5) 醫(yī)療模式的改變:相對(duì)比傳統(tǒng)生物制劑���,細(xì)胞 / 基因治療制品可能是每批都給病人單 獨(dú)生產(chǎn)和給藥,產(chǎn)品的批次和產(chǎn)量更為有限��。
此外��,罕見病用藥(生物制品居多)和細(xì)胞 / 基因治療產(chǎn)品在我國(guó)還面臨著巨大的進(jìn)口清 關(guān)檢驗(yàn)問題���。細(xì)胞 / 基因治療制品可能是每批都給病人單獨(dú)生產(chǎn)和給藥���。如果每批都需要預(yù)留 出進(jìn)口商檢放行的用量�����,則每個(gè)批次都需要在中國(guó)增加生產(chǎn)產(chǎn)量�����,這影響了生產(chǎn)周期�����。
同時(shí)部分細(xì)胞 / 基因治療產(chǎn)品可能需要基于患者個(gè)體指標(biāo)(如體重�����,AAV 滴度等)按需配藥���,并在短 時(shí)間內(nèi)送達(dá)至病入膏肓的患者用藥,而且此時(shí)病人通常都已病入膏肓��,因此對(duì)此類藥品進(jìn)口批 批商檢放行大大影響了患者用藥的時(shí)效性和可及性及成本�����,可能延遲病人獲得藥品的時(shí)間,也 導(dǎo)致病人的收益比大大降低�����,讓此類產(chǎn)品在中國(guó)很難被引入�����。
因此�����,很大程度上注冊(cè)檢驗(yàn)以及上市后的進(jìn)口檢驗(yàn)會(huì)很大程度的降低了行業(yè)進(jìn)行罕見病用 藥(包括細(xì)胞 / 基因治療)注冊(cè)的意愿:巨大額的進(jìn)口檢驗(yàn)用藥花費(fèi)�����,較長(zhǎng)的進(jìn)口檢驗(yàn)周期�����,相較于較短的產(chǎn)品效期無法滿足要求等等因素都極大的挫傷了這類產(chǎn)品進(jìn)入中國(guó)的積極性���。相較于傳統(tǒng)制品來說,注冊(cè)檢驗(yàn)和上市后的進(jìn)口商檢成為了這類產(chǎn)品全球同步研發(fā)注冊(cè)的一個(gè)較大的障礙(具體討論請(qǐng)參見本章節(jié)附件:IFPMIA 關(guān)于 ATMP 進(jìn)口檢驗(yàn)的建議函)。
• 制定新指南可以給工業(yè)界和監(jiān)管機(jī)構(gòu)帶來的益處:
通過基于產(chǎn)品的生產(chǎn)特性���,同時(shí)考慮全球比較統(tǒng)一的 GMP 規(guī)范和監(jiān)管要求���,單獨(dú)為此類 產(chǎn)品建立一個(gè)更加明確又相對(duì)靈活的注冊(cè)檢驗(yàn)啟動(dòng)流程,既可以在一定程度上減少行業(yè)上市前 的會(huì)議申請(qǐng)問詢��,減少藥審中心一些重復(fù)的工作量���,同時(shí)也便于行業(yè)在進(jìn)行商業(yè)可行性評(píng)價(jià)的 時(shí)候有更大的意愿將此類產(chǎn)品引入中國(guó)���,從而惠及更多的患者。
建議:
建議構(gòu)建合理的罕見病用藥檢驗(yàn)啟動(dòng)指南:可以根據(jù)批量及年產(chǎn)能力進(jìn)行區(qū)分�����。當(dāng)其低于某一水平時(shí)�����,可以僅進(jìn)行一個(gè)批次的注冊(cè)檢驗(yàn)和方法復(fù)核��。同時(shí)采用加強(qiáng)上市后 監(jiān)管的角度來豁免此類產(chǎn)品的上市后官方口岸檢驗(yàn)�����。
建議基于細(xì)胞/基因治療制品特點(diǎn)建立適合于該類產(chǎn)品的注冊(cè)檢驗(yàn)及進(jìn)口檢驗(yàn)的要求:
如酌情減免相應(yīng)的注冊(cè)檢驗(yàn)要求以及豁免上市后的進(jìn)口檢驗(yàn)。
修訂中檢院發(fā)布的藥品注迥檢驗(yàn)工作程序和技術(shù)要求
總結(jié):
注冊(cè)檢驗(yàn)是我國(guó)藥品注冊(cè)申請(qǐng)審評(píng)審批的重要環(huán)節(jié)���,對(duì)于保障用藥有重要意義��。當(dāng)前行業(yè)在做藥檢時(shí)發(fā)現(xiàn)缺少藥審中心與中檢院的溝通機(jī)制���,樣品要求與國(guó)際的工藝驗(yàn)證指導(dǎo)原則不一 致,推薦加急制定���。并向工業(yè)界提供相應(yīng)的培訓(xùn)�����。
差異分析成果:
該文件于 2020 年 7 月 1 日由中檢院發(fā)布�����,適用于藥品檢驗(yàn)機(jī)構(gòu)開展的,為支撐中藥�����、化學(xué)藥、 生物制品和按藥品管理的體外診斷試劑上市許可申請(qǐng)審評(píng)審批的樣品檢驗(yàn)和標(biāo)準(zhǔn)復(fù)核���,以及制劑審評(píng)需要的化學(xué)原料藥�����、生物制品原液���、藥用輔料、直接接觸藥品的包裝材料和容器的檢驗(yàn)�����。 但相對(duì)于歐美要求��,其存在如下區(qū)別:
1) 檢驗(yàn)類別的區(qū)別
歐美在臨床試驗(yàn)申請(qǐng)���、上市申請(qǐng)和重大藥學(xué)變更時(shí)��,不強(qiáng)制要求做注冊(cè)檢驗(yàn)(具體對(duì)標(biāo)表 格請(qǐng)參見啟動(dòng)注冊(cè)檢測(cè)的內(nèi)容);
2) 具體樣品要求的區(qū)別
a. 申請(qǐng)人在完成支持藥品上市的藥學(xué)相關(guān)研究���,確定質(zhì)量標(biāo)準(zhǔn),完成商業(yè)化規(guī)模生產(chǎn)工藝驗(yàn)證后�����,可向中國(guó)食品藥品檢定研究院(以下簡(jiǎn)稱中檢院)或省級(jí)藥品監(jiān)督管理 部門提出藥品注冊(cè)檢驗(yàn)申請(qǐng)。
b. 注冊(cè)檢驗(yàn)用樣品應(yīng)該為商業(yè)化規(guī)模生產(chǎn)的樣品�����,檢驗(yàn)樣品的相關(guān)信息(如產(chǎn)地���、直接接觸藥品的包裝材料等)應(yīng)與申請(qǐng)上市許可時(shí)提供的信息一致���。
c.(生物制品)成品為多種規(guī)格的,每個(gè)規(guī)格為三批樣品�����,成品處方相同���,存在有多種規(guī)格的�����,應(yīng)提供最大規(guī)格的三批樣品和其他規(guī)格至少一批樣品進(jìn)行檢驗(yàn)。
d.(化學(xué)藥品)樣品應(yīng)包裝完整���,有完整標(biāo)簽���,境內(nèi)藥品標(biāo)簽內(nèi)容應(yīng)符合國(guó)家藥品監(jiān) 督管理局藥品標(biāo)簽說明書相關(guān)文件規(guī)定�����,無正規(guī)標(biāo)簽的樣品���,必需貼有臨時(shí)標(biāo)簽; 標(biāo)簽內(nèi)容至少包括:檢品名稱、批號(hào)��、規(guī)格�����、生產(chǎn)單位;已確定效期的樣品標(biāo)簽上應(yīng)注明效期��,有特殊儲(chǔ)存條件要求的���,標(biāo)簽上需注明儲(chǔ)存條件���。境外已上市的制劑應(yīng)為完整市售包裝。抽樣樣品應(yīng)封簽完整無損,簽名或蓋章清晰可辨���。樣品標(biāo)簽內(nèi)容必須與資料相應(yīng)內(nèi)容一致��。
e.(化學(xué)藥品)(二)標(biāo)準(zhǔn)物質(zhì):提供檢驗(yàn)及方法學(xué)驗(yàn)證所涉及的標(biāo)準(zhǔn)物質(zhì)(對(duì)照品 或標(biāo)準(zhǔn)品)��,為滿足注冊(cè)檢驗(yàn)的 3 倍量���。標(biāo)準(zhǔn)物質(zhì)剩余有效期的要求與樣品一致。
f. 第三十四條 藥品注冊(cè)申請(qǐng)受理前及受理時(shí)啟動(dòng)的注冊(cè)檢驗(yàn)���,中藥���、化藥需要商業(yè)規(guī) 模生產(chǎn)的三批樣品,生物制品原則上需要商業(yè)規(guī)模連續(xù)生產(chǎn)的三批樣品�����,特殊情形 的除外���。(《藥品注冊(cè)核查檢驗(yàn)啟動(dòng)工作程序(試行)》)
• 中國(guó)實(shí)施實(shí)踐問題
1. 根據(jù)我國(guó)的監(jiān)管體系�����,藥審中心根據(jù)風(fēng)險(xiǎn)決定是否啟動(dòng)注冊(cè)檢驗(yàn)���,中檢院承擔(dān)注冊(cè)檢驗(yàn) 的實(shí)施工作。當(dāng)前未建立藥審中心和中檢院共同交流�����、達(dá)成一致的意見的渠道�����。存在的問題包 括(但不限于):
1) 注冊(cè)檢驗(yàn)用樣品的批次數(shù)���,藥審中心和中檢院的規(guī)范均有規(guī)定�����,行業(yè)根據(jù)產(chǎn)品特性 和 / 或上市后變更的影響程度擬建議樣品批次數(shù)���,需要有共同交流的渠道確定樣品 的批次數(shù)。注冊(cè)檢驗(yàn)用樣品準(zhǔn)備的周期較長(zhǎng)�����,尤其是生物制品,其從生產(chǎn)到放行需 要數(shù)月的時(shí)間���,因此���,需要在遞交上市申請(qǐng)或重大變更前確定檢驗(yàn)批次數(shù)。
2) 中檢院對(duì)申報(bào)的藥品質(zhì)量標(biāo)準(zhǔn)進(jìn)行復(fù)核并提出意見�����,但不修改申報(bào)質(zhì)量標(biāo)準(zhǔn)���。當(dāng)出 現(xiàn)注冊(cè)檢驗(yàn)相關(guān)問題時(shí)���,可能會(huì)引發(fā)二次藥檢,延緩病人得到藥品的時(shí)間和國(guó)家局 改革的初衷相悖��。
2. 注冊(cè)檢驗(yàn)要求完成商業(yè)規(guī)模生產(chǎn)工藝驗(yàn)證后方可進(jìn)行�����,并使用商業(yè)化規(guī)模生產(chǎn)的樣品��。 對(duì)于 ( 常規(guī)工藝 ) 生產(chǎn)的化藥���,國(guó)際上工藝驗(yàn)證的法規(guī)不強(qiáng)制要求所有類型的產(chǎn)品在上市申請(qǐng) 時(shí)完成生產(chǎn)規(guī)模的工藝驗(yàn)證(詳見批準(zhǔn)后生產(chǎn)條目下表 1)��。因此���,在全球同步研發(fā)���,( 常規(guī)工藝 ) 生產(chǎn)的化藥上市申請(qǐng)時(shí)���,完成商業(yè)化規(guī)模的工藝驗(yàn)證和提供商業(yè)化規(guī)模生產(chǎn)的 3 批樣品就成了挑戰(zhàn)�����。
3. 注冊(cè)檢驗(yàn)用樣品批數(shù)要求問題:
藥品注冊(cè)檢驗(yàn)工作程序和技術(shù)要求規(guī)范(試行)(2020 年版)規(guī)定:化學(xué)藥品的樣品為 多種規(guī)格的���,每個(gè)規(guī)格為三批樣品;僅對(duì)于液體制劑、半固體制劑(如軟膏��、乳膏等)�����,如處 方相同���,存在有多種規(guī)格的��,可根據(jù)具體情況確定一種規(guī)格的三批樣品和其他規(guī)格至少一批樣 品進(jìn)行檢驗(yàn)���。生物制品的成品為多種規(guī)格的�����,每個(gè)規(guī)格為三批樣品���,成品處方相同,存在有多 種規(guī)格的��,應(yīng)提供最大規(guī)格的三批樣品和其他規(guī)格至少一批樣品進(jìn)行檢驗(yàn)��。
藥品注冊(cè)核查檢驗(yàn)啟動(dòng)工作程序(試行)(2022 年發(fā)布)規(guī)定:第三十四條 藥品注冊(cè)申 請(qǐng)受理前及受理時(shí)啟動(dòng)的注冊(cè)檢驗(yàn)���,中藥��、化藥需要商業(yè)規(guī)模生產(chǎn)的三批樣品���,生物制品原則 上需要商業(yè)規(guī)模連續(xù)生產(chǎn)的三批樣品,特殊情形的除外�����。對(duì)于開展動(dòng)態(tài)生產(chǎn)現(xiàn)場(chǎng)核查的品種, 應(yīng)抽取動(dòng)態(tài)生產(chǎn)樣品���。
對(duì)于審評(píng)過程中提出的質(zhì)量標(biāo)準(zhǔn)單項(xiàng)或部分項(xiàng)目復(fù)核���,原則上需要三批樣品開展復(fù)核檢驗(yàn)。
根據(jù)歐美的工藝驗(yàn)證指導(dǎo)原則���,通常工藝驗(yàn)證需要連續(xù)生產(chǎn) 3 批,然而���,對(duì)于多規(guī)格產(chǎn)品�����,未要求所有的規(guī)格均需生產(chǎn) 3 批���。工藝驗(yàn)證可采用括號(hào)法(詳見下文,2��、工藝驗(yàn)證管理要求章節(jié))�����,驗(yàn)證條件應(yīng)覆蓋極端因素。因此���,上市申請(qǐng)時(shí)無法提供規(guī)范所要求的樣品批次數(shù)�����。
4. 上市后變更�����,由于產(chǎn)量的問題��,如罕見病藥物��,可能無法提供規(guī)范所要求的批次數(shù)���。
5. 化學(xué)藥品送檢樣品要求 MAH,包裝規(guī)格 , 包裝廠等所有信息均要求和擬定中國(guó)上市的一 樣 , 也不接受中國(guó)市場(chǎng)上實(shí)際上市的分包裝產(chǎn)品 , 這些信息的不同并不影響產(chǎn)品的質(zhì)量 , 但顯 著制約了可用于藥品檢驗(yàn)的產(chǎn)品的可及性���。由于法規(guī)的變化 , 將來越來多的新產(chǎn)品中國(guó)會(huì)和國(guó) 外同步遞交 , 要求相同 MAH 的市售包裝會(huì)顯著延緩新產(chǎn)品在中國(guó)的上市�����。
6. 化學(xué)藥品的對(duì)照品要求送檢時(shí)剩余效期不少于 180 個(gè)工作日���,對(duì)于很多不穩(wěn)定的對(duì)照品 , 或者復(fù)檢期只有一年的對(duì)照品 , 難以滿足此要求�����,建議有一定的靈活度�����。如行業(yè)可以在檢驗(yàn)的 時(shí)候提供對(duì)照品的復(fù)檢信息等�����。
7. 藥品注冊(cè)檢驗(yàn)包括樣品檢驗(yàn)和標(biāo)準(zhǔn)復(fù)核���。樣品檢驗(yàn) 60 個(gè)工作日��,樣品檢驗(yàn)和標(biāo)準(zhǔn)復(fù)核 90 個(gè)工作日���。對(duì)于上市后變更���,僅需樣品檢驗(yàn)���、無需標(biāo)準(zhǔn)復(fù)核的,有時(shí)會(huì)被要求按照注冊(cè)檢 驗(yàn)執(zhí)行(即同時(shí)包含了樣品檢驗(yàn)和標(biāo)準(zhǔn)復(fù)核)�����,檢驗(yàn)周期為 90 工作日�����,其對(duì)藥品和對(duì)照品的 剩余效期要求也會(huì)不同��。
8. 由于樣品要求復(fù)雜及準(zhǔn)備困難���,當(dāng)觸發(fā)二次藥檢時(shí)���,建議考慮可以接受使用藥檢留樣。
9. 當(dāng)前疫苗類產(chǎn)品在 CTA 階段仍需對(duì)原液和制劑進(jìn)行注冊(cè)檢驗(yàn)�����。CTA 階段生產(chǎn)工藝和質(zhì) 量標(biāo)準(zhǔn)和分析方法尚未最終建立�����,對(duì)該階段生產(chǎn)的樣品進(jìn)行注冊(cè)檢驗(yàn),意義有限�����。
• 相關(guān)的國(guó)內(nèi)監(jiān)管法規(guī) :
- 《藥品注冊(cè)管理辦法》
- 《藥品注冊(cè)核查檢驗(yàn)啟動(dòng)工作程序(試行)》
建議:
現(xiàn)行的文件無法解決在藥檢過程中行業(yè)遇到的所有問題�����,建議藥審中心和中檢院能協(xié)商并 共同出臺(tái)相關(guān)管理辦法���,去改善現(xiàn)有的流程:
1. 建議建立藥審中心���、中檢院和申請(qǐng)人針對(duì)注冊(cè)檢驗(yàn)共同交流,在某個(gè)時(shí)間節(jié)點(diǎn)���,達(dá)成一致意見的渠道�����,當(dāng)出現(xiàn)爭(zhēng)議時(shí)能共同決。現(xiàn)有時(shí)候會(huì)出現(xiàn)不同監(jiān)管方對(duì)同一要求或建議不統(tǒng)一�����, 行業(yè)無法理解如何按需求去改進(jìn)或提出異議。建議藥審中心和中檢院能有靈活的溝通流程和渠 道��,在雙方達(dá)成一致后��,再與行業(yè)商討�����。這個(gè)是急需建立的流程來保證藥品的可及性���。
2. 建議 ( 常規(guī)工藝 ) 生產(chǎn)的化學(xué)藥品的注冊(cè)檢驗(yàn)不要求商業(yè)規(guī)模工藝的樣品�����,提供的樣品 應(yīng)具有商業(yè)化規(guī)模的代表性��。
3. 建議不強(qiáng)制要求化學(xué)藥品提供相同 MAH 的市售包裝 , 提供的樣品質(zhì)量具有代表性即可���。 對(duì)照品不強(qiáng)制剩余效期 180 個(gè)工作日。
4. 上市申請(qǐng)時(shí)���,對(duì)于多規(guī)格產(chǎn)品��,建議根據(jù)工藝的風(fēng)險(xiǎn)(參考工藝驗(yàn)證的設(shè)計(jì))�����、產(chǎn)品的 包裝���、穩(wěn)定性行為等因素��,合理選擇樣品��。對(duì)于在一個(gè)或多個(gè)藥品生產(chǎn)場(chǎng)地生產(chǎn)藥物制劑�����,對(duì) 于存在多個(gè)原料藥 / 原液及制劑生產(chǎn)場(chǎng)地的���,建議接受上市許可人共提供 3 批樣品進(jìn)行藥品注 冊(cè)檢驗(yàn),其中至少包含每個(gè)場(chǎng)地生產(chǎn)的一批樣品���。
5. 對(duì)于上市后變更需要進(jìn)行注冊(cè)檢驗(yàn)的樣品批次選擇���,除考慮上述因素外,建議還應(yīng)考慮 以下(但不限于)因素:變更的風(fēng)險(xiǎn)(如原液產(chǎn)地變更不影響制劑質(zhì)量的��,可豁免制劑的注冊(cè) 檢驗(yàn))���,藥品的供應(yīng)情況(如罕見病藥物���,生產(chǎn)批次數(shù)較少),藥品歷史的檢驗(yàn)結(jié)果等���,允許 減少或豁免上市后變更的藥品檢驗(yàn)批次數(shù)���。
6. 對(duì)于疫苗產(chǎn)品,新《注冊(cè)管理辦法》以及中檢院《藥品注冊(cè)檢驗(yàn)工作程序和技術(shù)要求規(guī) 范(試行)》發(fā)布后�����,均未對(duì)疫苗 CTA 檢驗(yàn)以及臨床試驗(yàn)樣品檢驗(yàn)進(jìn)行要求���,但是實(shí)際申報(bào)過 程中��,仍要求在臨床試驗(yàn)啟動(dòng)前完成 3 批原液和 3 批制劑的注冊(cè)檢驗(yàn)�����,同時(shí)要求對(duì)臨床試驗(yàn)用 樣品進(jìn)行注冊(cè)檢驗(yàn)�����。然而進(jìn)口疫苗產(chǎn)品的實(shí)際操作中���,由于藥監(jiān)部門和海關(guān)之間沒有流暢的銜 接途徑,會(huì)導(dǎo)致樣品進(jìn)口通關(guān)��、送檢等流程受到很多阻礙�����。因此申請(qǐng)人在制訂項(xiàng)目計(jì)劃時(shí)�����,無 法準(zhǔn)確評(píng)估注冊(cè)檢驗(yàn)對(duì)項(xiàng)目進(jìn)度的影響�����,進(jìn)而極大的影響 MRCT 國(guó)內(nèi)部分的臨床試驗(yàn)的啟動(dòng)及 入組��,滯后創(chuàng)新藥的全球同步研發(fā)�����。因此,在疫苗產(chǎn)品臨床階段已有相應(yīng)的安全性監(jiān)控手段的 前提下�����,仍要求申請(qǐng)人在臨床試驗(yàn)啟動(dòng)前額外完成 3 批原液和 3 批制劑的注冊(cè)檢驗(yàn)��,并不能提 供額外的風(fēng)險(xiǎn)控制價(jià)值���。因此,基于風(fēng)險(xiǎn)可控的原則�����,并支持產(chǎn)品的加速上市���,建議不再強(qiáng)制 要求疫苗類產(chǎn)品在臨床試驗(yàn)開展前完成注冊(cè)檢驗(yàn)��,允許行業(yè)在臨床試驗(yàn)過程中完成注冊(cè)檢驗(yàn)���。
參考文獻(xiàn)
1.國(guó)內(nèi)外藥品技術(shù)指導(dǎo)原則體系對(duì)比研究 (藥學(xué)部分》,國(guó)家藥品監(jiān)督管理局藥品審評(píng)中心�����,中國(guó)藥品監(jiān)督管理研究會(huì),藥品監(jiān)管研究國(guó)際交流專業(yè)委員會(huì)��,中國(guó)外商投資企業(yè)協(xié)會(huì)藥品研制和開發(fā)工作委員會(huì)(RDPAC)���,2022年11月