摘 要 Abstract
本文梳理了醫(yī)療器械創(chuàng)新產(chǎn)品注冊(cè)程序?qū)嵤┮詠?lái),在我國(guó)上市的24 個(gè)進(jìn)口創(chuàng)新醫(yī)療器械的時(shí)間����,發(fā)現(xiàn)與美國(guó)上市的時(shí)間差平均為4.4 年,這與國(guó)家提倡的盡快引入先進(jìn)產(chǎn)品�����、鼓勵(lì)全球同步上市的目標(biāo)尚存在差距��。為研究進(jìn)口創(chuàng)新醫(yī)療器械在我國(guó)同步上市的影響因素��,本文整理了我國(guó)創(chuàng)新醫(yī)療器械資質(zhì)申請(qǐng)��、臨床試驗(yàn)備案和審批以及注冊(cè)申請(qǐng)的流程與要求����,并與美國(guó)突破性醫(yī)療器械的上市路徑進(jìn)行對(duì)比。結(jié)果表明����,專利申請(qǐng)��、型式檢驗(yàn)以及臨床試驗(yàn)審批是最易導(dǎo)致上市時(shí)間滯后的環(huán)節(jié)�����,進(jìn)而影響我國(guó)加入全球多中心臨床試驗(yàn)和同步研發(fā)��。本文提出延長(zhǎng)創(chuàng)新醫(yī)療器械資質(zhì)時(shí)長(zhǎng)����、規(guī)范醫(yī)療器械臨床階段變更路徑�����、統(tǒng)一臨床試驗(yàn)審批中對(duì)境內(nèi)外企業(yè)的要求��、加快國(guó)際標(biāo)準(zhǔn)本地轉(zhuǎn)化����、完善專利制度、允許創(chuàng)新產(chǎn)品自主靈活定價(jià)����、鼓勵(lì)境內(nèi)企業(yè)開展海外合作等幾點(diǎn)建議��,以期為進(jìn)口創(chuàng)新醫(yī)療器械在我國(guó)同步上市政策研究提供思路與借鑒��。
This paper reviews the time of 24 imported innovative medical devices approved in China since the implementation of the innovative medical devices registration procedure, and finds the average time lag is 4.4 years compared to their market launch in the United States, indicating a gap from the national advocacy of promptly introducing advanced products and encouraging global simultaneous market launches. To investigate the factors affecting the simultaneous market entry of imported innovative medical devices in China, this article outlines the processes and requirements for innovative medical device designation application, clinical trial filing and approval, and market application for innovative medical devices. A comparison is made with the marketing pathway for breakthrough medical devices in the United States. The comparison reveals that patent application, type testing, and clinical trial approval are the key stages that most likely contribute to the time lag in market approval, impeding China's participation in global multicenter clinical trials and collaborative research. Therefore, this article proposes relevant policy considerations and recommendations, including extending the designation validity duration of innovative medical devices, standardizing the pathways for device changes during the clinical stage, unifying the requirements for domestic and foreign applicants in clinical trial approvals, accelerating the local translation of international standards, improving the patent system, allowing innovative products set prices independently, and encouraging domestic enterprises to engage in overseas collaborations in multiple ways.
關(guān)鍵詞 Key words
創(chuàng)新醫(yī)療器械�����;同步上市;臨床試驗(yàn)審批����;型式檢驗(yàn);突破性醫(yī)療器械
innovative medical device; simultaneous marketing; clinical trial approval; type testing;breakthrough device
深化醫(yī)藥衛(wèi)生體制改革是建設(shè)健康中國(guó)行動(dòng)的重點(diǎn)內(nèi)容����,其中加速藥品和醫(yī)療器械的審批上市是建設(shè)健康中國(guó)、深化醫(yī)療衛(wèi)生領(lǐng)域供給側(cè)結(jié)構(gòu)性改革的重要舉措��。促進(jìn)以臨床需求為導(dǎo)向的創(chuàng)新藥品和醫(yī)療器械在我國(guó)上市�����,有利于擴(kuò)充現(xiàn)有診療手段����,豐富治療方案����?����!吨腥A人民共和國(guó)國(guó)民經(jīng)濟(jì)和社會(huì)發(fā)展第十四個(gè)五年規(guī)劃和2035 年遠(yuǎn)景目標(biāo)綱要》中����,明確提出了“完善創(chuàng)新藥物、疫苗����、醫(yī)療器械等快速審評(píng)審批機(jī)制,加快臨床急需和罕見病治療藥品����、醫(yī)療器械審評(píng)審批”[1],強(qiáng)調(diào)了對(duì)技術(shù)創(chuàng)新產(chǎn)品和臨床急需產(chǎn)品的特別關(guān)注�����。在《“十四五”醫(yī)藥工業(yè)發(fā)展規(guī)劃》中��,進(jìn)一步對(duì)醫(yī)藥產(chǎn)業(yè)的創(chuàng)新能力及國(guó)際化地位進(jìn)行了展望,提出要吸引全球醫(yī)藥創(chuàng)新要素向國(guó)內(nèi)集聚�����,包括吸引全球創(chuàng)新藥品和醫(yī)療器械率先在我國(guó)注冊(cè)��,整體縮短創(chuàng)新產(chǎn)品國(guó)內(nèi)外上市時(shí)間差�����,促進(jìn)臨床急需的境外已上市新藥和醫(yī)療器械盡快在境內(nèi)注冊(cè)[2]��。2021 年����,國(guó)家藥監(jiān)局等8 部門聯(lián)合印發(fā)的《“十四五”國(guó)家藥品安全及促進(jìn)高質(zhì)量發(fā)展規(guī)劃》聚焦在產(chǎn)品審評(píng)審批的目標(biāo)上��,提出“加強(qiáng)創(chuàng)新產(chǎn)品審評(píng)能力�����,能夠同步審評(píng)審批全球創(chuàng)新藥物和醫(yī)療器械�����,支持境外新藥和醫(yī)療器械在境內(nèi)同步上市”[3]。由此可知��,如何使我國(guó)公眾盡早用上國(guó)際先進(jìn)醫(yī)療產(chǎn)品已成為我國(guó)藥品監(jiān)管部門關(guān)注的重點(diǎn)之一�����。
讓國(guó)際先進(jìn)醫(yī)藥產(chǎn)品在我國(guó)同步上市對(duì)于醫(yī)藥行業(yè)全鏈條的發(fā)展都具有促進(jìn)意義:①滿足臨床需求����,使患者不必萬(wàn)里求醫(yī),可同步分享全球醫(yī)藥創(chuàng)新成果��;②吸引全球醫(yī)療資源向我國(guó)匯集����,提高我國(guó)患者和市場(chǎng)的關(guān)注度;③彌補(bǔ)診療空白��,學(xué)習(xí)國(guó)際最先進(jìn)的治療方案�����,提高我國(guó)醫(yī)藥從業(yè)者在國(guó)際上的話語(yǔ)權(quán)和參與度��;④提高監(jiān)管機(jī)構(gòu)的創(chuàng)新產(chǎn)品評(píng)價(jià)能力��,逐漸與國(guó)際監(jiān)管、標(biāo)準(zhǔn)接軌����;⑤提高市場(chǎng)活力,以進(jìn)口競(jìng)爭(zhēng)帶動(dòng)我國(guó)醫(yī)藥產(chǎn)品研發(fā)水平進(jìn)步��,促進(jìn)全球技術(shù)交流����,有利于國(guó)產(chǎn)產(chǎn)品走出國(guó)門。
目前�����,我國(guó)藥品領(lǐng)域已有產(chǎn)品基本實(shí)現(xiàn)了全球同步上市��,例如勃林格殷格翰公司研發(fā)的佩索利單抗注射液和艾伯維公司研發(fā)的烏帕替尼緩釋片��,我國(guó)與美國(guó)同步研發(fā)����,并參與國(guó)際多中心臨床試驗(yàn)����,實(shí)現(xiàn)了同步申報(bào)以及同步上市��。然而��,進(jìn)口醫(yī)療器械領(lǐng)域目前罕有同步上市情況�����。因此�����,本文探討了進(jìn)口醫(yī)療器械全球同步上市現(xiàn)狀�����,以期探究影響我國(guó)進(jìn)口醫(yī)療器械同步上市的因素��,并提出相應(yīng)政策建議�����。
1����、 同步上市的路徑與現(xiàn)狀
關(guān)于同步上市的定義在我國(guó)現(xiàn)有法規(guī)中尚未有明確的表述�����,一般可以理解為產(chǎn)品在我國(guó)境內(nèi)和境外的上市時(shí)間差盡可能小?����?紤]到各國(guó)和地區(qū)審評(píng)時(shí)限不同��,同步申報(bào)也可以劃入同步上市的范疇��。
若要縮短進(jìn)口產(chǎn)品上市的“時(shí)間差”甚至是爭(zhēng)取同步上市����,就必須利用好特殊注冊(cè)程序?�!夺t(yī)療器械注冊(cè)與備案管理辦法》中�����,規(guī)定了3 種特殊審批程序可以加速醫(yī)療器械的上市審批��。3 種路徑的適用場(chǎng)景各有側(cè)重:①創(chuàng)新產(chǎn)品注冊(cè)程序��,側(cè)重對(duì)有核心技術(shù)發(fā)明專利����、有顯著臨床優(yōu)勢(shì)且國(guó)際領(lǐng)先的首創(chuàng)產(chǎn)品的扶持。②優(yōu)先注冊(cè)程序����,側(cè)重臨床急需、國(guó)家研發(fā)重點(diǎn)及其他情形下的優(yōu)先審查��。③應(yīng)急注冊(cè)程序��,側(cè)重對(duì)突發(fā)公共衛(wèi)生事件的應(yīng)對(duì)[4]����。
值得注意的是,《醫(yī)療器械監(jiān)督管理?xiàng)l例》第十六條規(guī)定“未在境外上市的創(chuàng)新醫(yī)療器械��,可以不提交注冊(cè)申請(qǐng)人所在國(guó)(地區(qū))主管部門準(zhǔn)許該醫(yī)療器械上市銷售的證明文件”[5]�����。也就是說��,其他途徑都需要該產(chǎn)品在原產(chǎn)國(guó)(地區(qū))獲批后����,將原產(chǎn)國(guó)(地區(qū))上市批件作為在境內(nèi)申報(bào)的遞交文件之一����,只有創(chuàng)新醫(yī)療器械最可能實(shí)現(xiàn)境內(nèi)外同步上市����。因此,創(chuàng)新醫(yī)療器械的申報(bào)路徑為本研究的重點(diǎn)����。
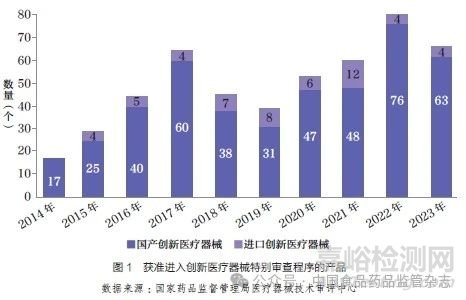
截至2023 年, 我國(guó)共有499 個(gè)產(chǎn)品獲準(zhǔn)進(jìn)入創(chuàng)新醫(yī)療器械特別審查程序�����,其中54 個(gè)為進(jìn)口產(chǎn)品����,約占10.8%,如圖1所示��。
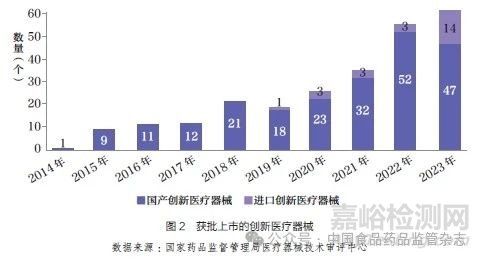
按照“標(biāo)準(zhǔn)不降低����,程序不減少”的原則,國(guó)家藥品監(jiān)督管理局醫(yī)療器械技術(shù)審評(píng)中心(以下簡(jiǎn)稱器審中心)對(duì)創(chuàng)新醫(yī)療器械產(chǎn)品給予了早期介入��、專人負(fù)責(zé)�����、加強(qiáng)溝通等幫助��,較其他第三類醫(yī)療器械首次注冊(cè)的平均時(shí)間縮短近3 個(gè)月[6]�����。截至2023 年�����,獲得創(chuàng)新醫(yī)療器械資質(zhì)的產(chǎn)品中��,已有250 個(gè)產(chǎn)品獲批����,其中進(jìn)口醫(yī)療器械24 個(gè),約占9.6%��,如圖2 所示�����。
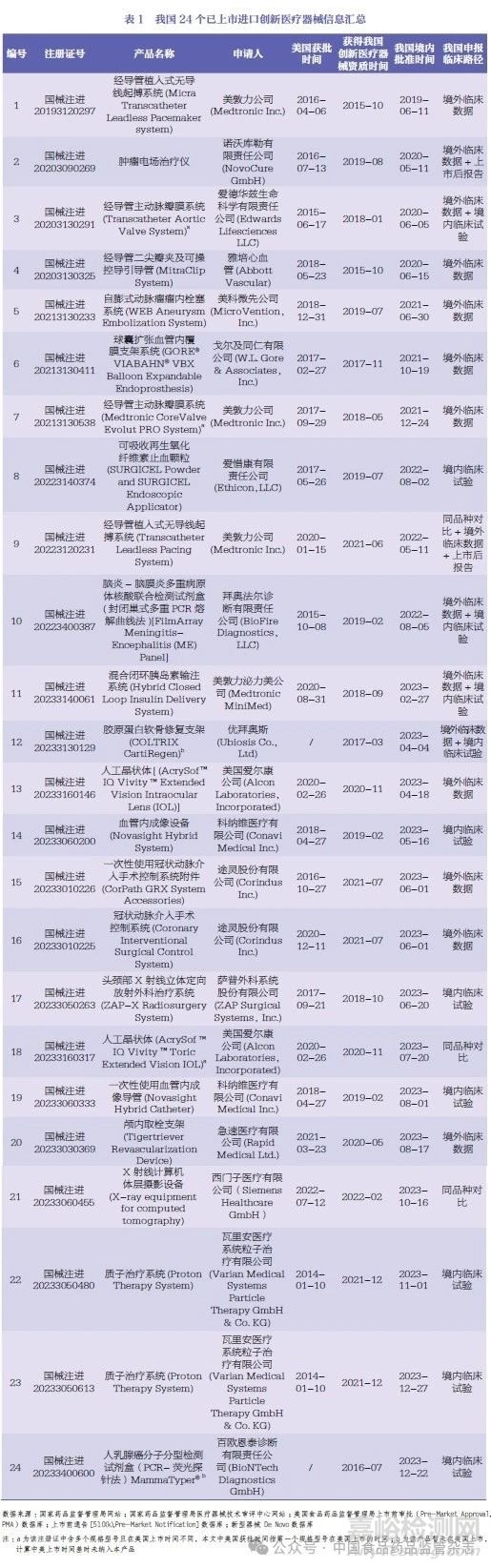
在已上市的24 個(gè)進(jìn)口創(chuàng)新醫(yī)療器械中,與美國(guó)上市時(shí)間差最短的是西門子醫(yī)療有限公司的X射線計(jì)算機(jī)體層攝影設(shè)備�����,在美國(guó)上市461 天后在我國(guó)上市�����。這24 個(gè)進(jìn)口產(chǎn)品中有22 個(gè)已在美國(guó)上市��,我國(guó)與美國(guó)上市的平均時(shí)間差是4.4 年����,與《“十四五”國(guó)家藥品安全及促進(jìn)高質(zhì)量發(fā)展規(guī)劃》中提出的境內(nèi)外同步上市的目標(biāo)尚存在差距。
本文匯總了我國(guó)已上市的24 個(gè)進(jìn)口創(chuàng)新醫(yī)療器械產(chǎn)品的基本信息以及在美國(guó)的獲批時(shí)間��,見表1��。需要說明的是��,這24 個(gè)產(chǎn)品不一定在美國(guó)首先上市�����,可能選擇了在歐盟、研發(fā)地或生產(chǎn)地申請(qǐng)上市��。本文只列舉了這24 個(gè)產(chǎn)品在美國(guó)的上市時(shí)間��。一是因?yàn)槊绹?guó)作為全球醫(yī)療器械的主要市場(chǎng)��,產(chǎn)品獲得FDA的批準(zhǔn)具有參考意義����。二是筆者希望通過與美國(guó)突破性醫(yī)療器械程序(Breakthrough Medical Device Program) 進(jìn)行對(duì)比�����,分析其優(yōu)勢(shì)����,以獲得提升我國(guó)相關(guān)規(guī)定的啟示。
2�����、 上市流程的挑戰(zhàn)
本文梳理了進(jìn)口創(chuàng)新醫(yī)療器械在我國(guó)上市的全部流程��,包括資質(zhì)申請(qǐng)�����、臨床路徑以及上市申報(bào)遞交流程,并從法規(guī)層面逐個(gè)分析了影響進(jìn)口創(chuàng)新醫(yī)療器械同步上市的因素����。同時(shí),對(duì)比美國(guó)食品藥品監(jiān)督管理局(Food and Drug Administration�����,F(xiàn)DA)的相應(yīng)程序�����,分析了同一產(chǎn)品如果在我國(guó)和美國(guó)同時(shí)啟動(dòng)能否達(dá)到相近的上市時(shí)間��,剖析我國(guó)與美國(guó)法規(guī)的差異��,并借鑒FDA 控制產(chǎn)品風(fēng)險(xiǎn)的方法��,提出了相應(yīng)政策建議����,以期為我國(guó)創(chuàng)新醫(yī)療器械加速上市提供借鑒與思路。
2.1 創(chuàng)新醫(yī)療器械申請(qǐng)
2014 年�����,《創(chuàng)新醫(yī)療器械特別審批程序(試行)》[7] 發(fā)布實(shí)施。為進(jìn)一步規(guī)范創(chuàng)新醫(yī)療器械的申報(bào)文件和流程����,國(guó)家藥品監(jiān)管部門又于2016 年發(fā)布了《創(chuàng)新醫(yī)療器械特別審批申報(bào)資料編寫指南》[8]。2018 年����,國(guó)家藥品監(jiān)管部門對(duì)上述2 個(gè)文件進(jìn)行了制修訂�����,并發(fā)布了《醫(yī)療器械技術(shù)審評(píng)中心創(chuàng)新醫(yī)療器械特別審查申請(qǐng)審查操作規(guī)范》[9]��,目的是更好地扶持創(chuàng)新產(chǎn)品�����,加快產(chǎn)品上市�����。
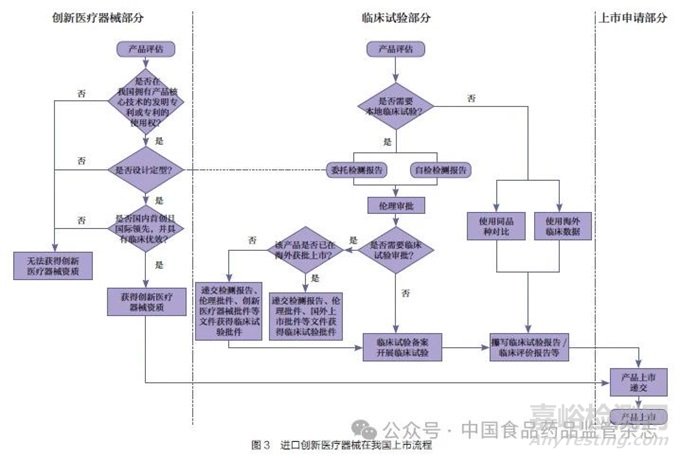
創(chuàng)新醫(yī)療器械需要符合以下3 個(gè)要求:①產(chǎn)品的創(chuàng)新性��。這需要申請(qǐng)人擁有核心技術(shù)的發(fā)明專利、依法獲得該專利的所有權(quán)或使用權(quán)為證明依據(jù)��;對(duì)于尚未在我國(guó)授權(quán)的專利�����,需提供檢索報(bào)告并以足夠的新穎性和創(chuàng)新性證明該專利未來(lái)可以獲得授權(quán)����。②該產(chǎn)品應(yīng)已基本定型,以關(guān)鍵性能的驗(yàn)證報(bào)告為證明�����。③該產(chǎn)品應(yīng)具有首創(chuàng)性和顯著的臨床優(yōu)效性��,與同類產(chǎn)品比較時(shí)應(yīng)有根本性的改變��,且技術(shù)處于領(lǐng)先地位����。
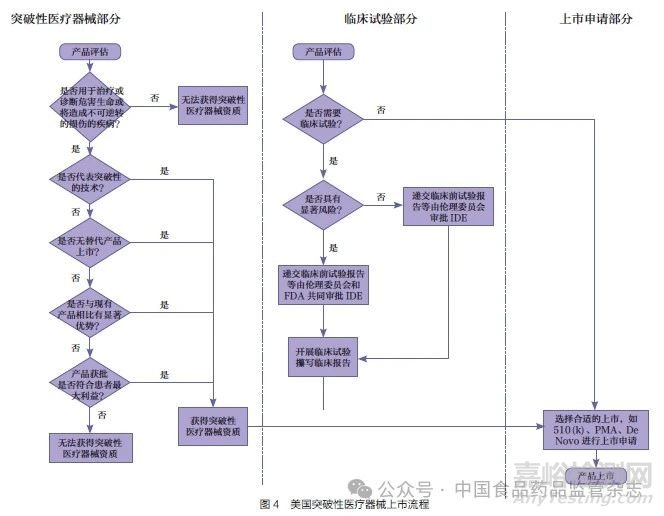
FDA 對(duì)于突破性醫(yī)療器械資質(zhì)的考察集中在產(chǎn)品的創(chuàng)新性以及臨床優(yōu)效性上。首要條件為該醫(yī)療器械為危及生命或?qū)⒃斐刹豢膳まD(zhuǎn)的損傷的疾病提供了更有效的診療手段��。另外����,突破性醫(yī)療器械應(yīng)滿足以下4 個(gè)次要條件中的至少1 個(gè):①代表突破性技術(shù)����;②沒有替代產(chǎn)品上市��;③相比現(xiàn)有產(chǎn)品具有顯著優(yōu)勢(shì)��;④該產(chǎn)品的獲批符合患者的最佳利益[10]�����。
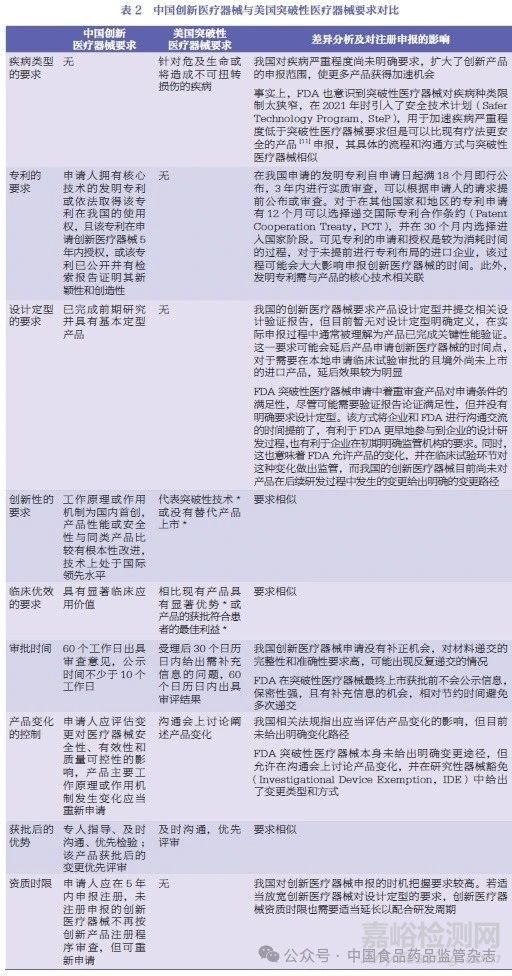
需要特別說明的是����,并不是只有在美國(guó)獲得了突破性療法的產(chǎn)品才能在我國(guó)獲批創(chuàng)新醫(yī)療器械��,在評(píng)估產(chǎn)品是否有替代產(chǎn)品以及與現(xiàn)有療法相比是否有顯著的臨床優(yōu)勢(shì)時(shí)��,都是針對(duì)本國(guó)(地區(qū))市場(chǎng)而言�����,因此二者并不一定是一一對(duì)應(yīng)的關(guān)系��。本文將我國(guó)創(chuàng)新醫(yī)療器械與美國(guó)突破性醫(yī)療器械的要求進(jìn)行了對(duì)比,總結(jié)了法規(guī)上的啟示并分析了出現(xiàn)上市時(shí)間差的原因��,見表2����。
2.2 臨床路徑的選擇
臨床評(píng)價(jià)路徑的選擇分為免于臨床評(píng)價(jià)、通過同品種醫(yī)療器械評(píng)價(jià)和通過臨床試驗(yàn)數(shù)據(jù)評(píng)價(jià)����。創(chuàng)新醫(yī)療器械具有強(qiáng)創(chuàng)新性,通常不滿足免于臨床評(píng)價(jià)的要求�����。截至2023 年已獲批且有公開審評(píng)報(bào)告的創(chuàng)新產(chǎn)品中�����,11.2% 的產(chǎn)品采用了同品種的評(píng)價(jià)方式�����,82.7% 的產(chǎn)品采用了臨床試驗(yàn)�����,6.1% 的產(chǎn)品采用以上兩種混合方式的臨床路徑完成了產(chǎn)品安全性和有效性的論證。其中��,采用臨床試驗(yàn)的數(shù)據(jù)來(lái)源既可以是本地臨床試驗(yàn)��,也可以是境外臨床試驗(yàn)����。
近年來(lái),為加快醫(yī)療器械上市步伐�����,減輕企業(yè)負(fù)擔(dān)����,監(jiān)管部門出臺(tái)了多項(xiàng)靈活政策,其中包括利用真實(shí)世界研究進(jìn)行臨床評(píng)價(jià)��。真實(shí)世界研究的數(shù)據(jù)可以是來(lái)自境外已上市國(guó)家和地區(qū)����,用于對(duì)我國(guó)人種外推或臨床條件的補(bǔ)充論證����,也可以是來(lái)自海南博鰲的特許使用��, 用于支持產(chǎn)品的申報(bào)注冊(cè)�����。目前����, 美國(guó)艾爾建公司的青光眼引流管XEN Glaucoma Treatment System 和美國(guó)強(qiáng)生眼力健公司的“Catalys 白力士”眼科飛秒激光治療機(jī)Catalys Precision Laser System 已成功利用樂城真實(shí)世界數(shù)據(jù)獲批��。前者從申請(qǐng)到獲批僅歷時(shí)5 個(gè)月�����,后者從首例手術(shù)到獲批也僅歷時(shí)14 個(gè)月����,驗(yàn)證了真實(shí)世界數(shù)據(jù)在臨床評(píng)價(jià)中的可行性,并加速了產(chǎn)品上市進(jìn)程�����。根據(jù)《關(guān)于支持建設(shè)博鰲樂城國(guó)際醫(yī)療旅游先行區(qū)的實(shí)施方案》[12]����,博鰲樂城先行區(qū)內(nèi)允許使用“境外已上市但境內(nèi)未上市”的產(chǎn)品�����,境外真實(shí)世界數(shù)據(jù)也只能在產(chǎn)品境外上市后獲得��,因此這兩種方式雖然大大縮短了審評(píng)審批時(shí)間����,也尚無(wú)法通過這種方式達(dá)到同步上市的目標(biāo)����。
我國(guó)規(guī)定在境內(nèi)開展臨床試驗(yàn)的產(chǎn)品需要提供檢測(cè)報(bào)告用于倫理審批。對(duì)于需要臨床試驗(yàn)審批的第三類高風(fēng)險(xiǎn)產(chǎn)品��,還需在臨床備案前獲得臨床試驗(yàn)批件��;未在境外上市的進(jìn)口產(chǎn)品��,需在臨床試驗(yàn)審批中提交創(chuàng)新批件�����。檢測(cè)報(bào)告可以是申請(qǐng)人出具的自檢報(bào)告或者有資質(zhì)的醫(yī)療器械檢驗(yàn)機(jī)構(gòu)出具的檢測(cè)報(bào)告�����。FDA對(duì)開展臨床的審批流程與我國(guó)類似��, 申請(qǐng)人需準(zhǔn)備研究性器械豁免(investigational device exemption����,IDE)的遞交。對(duì)于非顯著高風(fēng)險(xiǎn)(non-significant risk����,NSR) 的產(chǎn)品,IDE 僅需倫理審批����;對(duì)于顯著高風(fēng)險(xiǎn)(significant risk,SR)的產(chǎn)品��,還需要FDA 審批IDE 后才能開展臨床試驗(yàn)[13]����。為此,本文對(duì)我國(guó)和美國(guó)的臨床審批流程進(jìn)行了詳細(xì)對(duì)比�����,見表3。
2.3 上市遞交的要求
完成臨床試驗(yàn)并撰寫遞交所需相關(guān)資料后����,即可進(jìn)行上市申請(qǐng)遞交。我國(guó)與美國(guó)創(chuàng)新產(chǎn)品的上市路徑及流程類似�����,審評(píng)時(shí)限略有不同��,具體對(duì)比見表4����。
2.4 上市流程總結(jié)
圖3 和圖4 總結(jié)了我國(guó)和美國(guó)創(chuàng)新產(chǎn)品上市的流程。從流程圖上可以看出����,美國(guó)的突破性醫(yī)療器械資質(zhì)申請(qǐng)和臨床試驗(yàn)申請(qǐng)是兩個(gè)相對(duì)獨(dú)立的流程����,可以同時(shí)進(jìn)行。我國(guó)對(duì)于無(wú)需進(jìn)行臨床試驗(yàn)審批的產(chǎn)品�����,創(chuàng)新醫(yī)療器械的申請(qǐng)和臨床試驗(yàn)也可以同步進(jìn)行,但對(duì)于需要臨床試驗(yàn)審批且尚未在境外上市的產(chǎn)品�����,則需要先后進(jìn)行�����。根據(jù)《關(guān)于公布醫(yī)療器械注冊(cè)申報(bào)資料要求和批準(zhǔn)證明文件格式的公告》附件8 中對(duì)臨床試驗(yàn)審批資料的要求�����,境外申請(qǐng)人應(yīng)提交境外上市批件或創(chuàng)新批件[24]����。而創(chuàng)新批件的申請(qǐng)又要求產(chǎn)品已定型��,也就是說同一款第三類高風(fēng)險(xiǎn)產(chǎn)品如果在我國(guó)和美國(guó)同時(shí)啟動(dòng)����,我國(guó)在流程上可能會(huì)落后2~3 個(gè)時(shí)間節(jié)點(diǎn)。
3����、 政策思考
近些年����,國(guó)家藥品監(jiān)管部門出臺(tái)了多項(xiàng)政策加速進(jìn)口醫(yī)療器械的上市��,包括《接受醫(yī)療器械境外臨床試驗(yàn)數(shù)據(jù)技術(shù)指導(dǎo)原則》[25]提出可以利用境外數(shù)據(jù)作為臨床評(píng)價(jià)證據(jù)��,減少我國(guó)境內(nèi)的臨床試驗(yàn)數(shù)據(jù)規(guī)模;《真實(shí)世界數(shù)據(jù)用于醫(yī)療器械臨床評(píng)價(jià)技術(shù)指導(dǎo)原則(試行)》[26] 規(guī)范和引導(dǎo)了利用真實(shí)世界研究數(shù)據(jù)用于醫(yī)療器械臨床評(píng)價(jià)�����,并已在海南博鰲成功試點(diǎn)�����;新修訂《醫(yī)療器械監(jiān)督管理?xiàng)l例》[5] 明確了創(chuàng)新產(chǎn)品可以豁免境外上市批件,進(jìn)一步提前了進(jìn)口產(chǎn)品可進(jìn)行申報(bào)的時(shí)間點(diǎn)����。這些政策有效縮短了我國(guó)進(jìn)口產(chǎn)品與境外上市的時(shí)間差����。為實(shí)現(xiàn)“同步上市”����,筆者探索性地提出幾點(diǎn)建議��,以期進(jìn)一步縮短進(jìn)口醫(yī)療器械上市時(shí)間差。
3.1 適當(dāng)延長(zhǎng)創(chuàng)新醫(yī)療器械資格年限����,明確臨床試驗(yàn)階段的變更路徑,控制產(chǎn)品變更風(fēng)險(xiǎn)
我國(guó)申報(bào)創(chuàng)新醫(yī)療器械時(shí)要求產(chǎn)品設(shè)計(jì)定型�����,但后續(xù)臨床環(huán)節(jié)仍可能發(fā)生產(chǎn)品變更�����,這意味著產(chǎn)品可能需要重新進(jìn)行部分驗(yàn)證��、檢測(cè)甚至修改臨床方案��,因此建議可適當(dāng)放寬創(chuàng)新醫(yī)療器械5 年的資格期限����,使企業(yè)能更充分地進(jìn)行研發(fā)����,也避免重復(fù)審評(píng)創(chuàng)新申請(qǐng)��。
同時(shí)��,為應(yīng)對(duì)申報(bào)創(chuàng)新或臨床試驗(yàn)備案����、審批后發(fā)生的變化,建議監(jiān)管部門應(yīng)明確變更路徑����。目前�����,企業(yè)主要的應(yīng)對(duì)方式是與相關(guān)部門召開溝通咨詢會(huì)或者發(fā)文咨詢����,尚沒有明確的法規(guī)、指南等詳細(xì)說明變更路徑要求����。2022 年國(guó)家藥品監(jiān)督管理局藥品審評(píng)中心發(fā)布了《藥物臨床試驗(yàn)期間方案變更技術(shù)指導(dǎo)原則(試行)》�����,其參考了國(guó)際上對(duì)藥品臨床階段發(fā)生變更情況的要求,按照變更是否為實(shí)質(zhì)性變更��,以及實(shí)質(zhì)性變更是否會(huì)影響受試者安全等因素給出了3 條變更路徑[27]��。因此�����,建議器審中心也可以參考FDA 的IDE 變更路徑�����,將常見的變更情況分類規(guī)范�����,填補(bǔ)醫(yī)療器械臨床階段產(chǎn)品變更的規(guī)范的不足�����,明確每類變更路線所需的資料和審評(píng)時(shí)間�����,實(shí)現(xiàn)產(chǎn)品的全生命周期監(jiān)管�����,引導(dǎo)企業(yè)及時(shí)溝通�����、規(guī)范申報(bào)����,避免影響受試者安全,對(duì)于加入國(guó)際多中心臨床試驗(yàn)的企業(yè)還可以更好地協(xié)調(diào)境外臨床的時(shí)間����。
3.2 統(tǒng)一臨床試驗(yàn)審批中對(duì)境內(nèi)和境外申請(qǐng)人的要求��,取消進(jìn)口產(chǎn)品提交上市證明或創(chuàng)新醫(yī)療器械批件的要求
《需進(jìn)行臨床試驗(yàn)審批的第三類醫(yī)療器械目錄(2020 年修訂版)》中,對(duì)應(yīng)進(jìn)行臨床試驗(yàn)審批對(duì)象的要求為“與境內(nèi)外已上市產(chǎn)品相比����,采用全新設(shè)計(jì)、材料或機(jī)理����,和/ 或適用于全新適用范圍”[14]��,也就是說��,需要申報(bào)臨床試驗(yàn)審批的產(chǎn)品在某一方面是沒有可替代已獲批產(chǎn)品的����,即便并非為創(chuàng)新醫(yī)療器械��,也可以填補(bǔ)市場(chǎng)空白。在該背景下��,建議可適當(dāng)考慮優(yōu)先患者利益��,及時(shí)將新產(chǎn)品引入市場(chǎng)。盡管相關(guān)政策為我國(guó)企業(yè)提供了一定的政策傾斜��,但是臨床試驗(yàn)審批的目的是考察產(chǎn)品進(jìn)入臨床試驗(yàn)的安全性����。因此��,筆者認(rèn)為只要進(jìn)口產(chǎn)品可以提供證明臨床風(fēng)險(xiǎn)合理可控的證據(jù)��,并非必須通過境外上市論證臨床安全性才可以開展臨床試驗(yàn)��。同時(shí),進(jìn)口醫(yī)療器械的快速引入也有利于促進(jìn)國(guó)內(nèi)企業(yè)的研發(fā)和創(chuàng)新�����。
3.3 促進(jìn)國(guó)際標(biāo)準(zhǔn)本地轉(zhuǎn)化和更新,統(tǒng)一國(guó)內(nèi)外審評(píng)尺度
《國(guó)家藥品監(jiān)督管理局 國(guó)家標(biāo)準(zhǔn)化管理委員會(huì)關(guān)于進(jìn)一步促進(jìn)醫(yī)療器械標(biāo)準(zhǔn)化工作高質(zhì)量發(fā)展的意見》提出:“建立與國(guó)際標(biāo)準(zhǔn)快速聯(lián)動(dòng)的標(biāo)準(zhǔn)更新機(jī)制����,探索國(guó)內(nèi)標(biāo)準(zhǔn)與國(guó)際標(biāo)準(zhǔn)同步立項(xiàng),縮短國(guó)際標(biāo)準(zhǔn)轉(zhuǎn)化周期����。”[28] 目前����,我國(guó)醫(yī)療器械國(guó)際標(biāo)準(zhǔn)轉(zhuǎn)化率已達(dá)90% 以上����,其中最新版的轉(zhuǎn)化標(biāo)準(zhǔn)占全部的53%[29]����。及時(shí)轉(zhuǎn)化國(guó)際標(biāo)準(zhǔn)����,可以避免同一產(chǎn)品按照不同的標(biāo)準(zhǔn)和評(píng)價(jià)體系試驗(yàn)�����,對(duì)促進(jìn)醫(yī)療器械貿(mào)易和國(guó)際化監(jiān)管都有積極作用����。此外��,政策的差異帶來(lái)的影響是雙向的��,即進(jìn)口創(chuàng)新產(chǎn)品難以在我國(guó)同步上市��,我國(guó)出口的高價(jià)值醫(yī)療器械又相對(duì)較少����。除了需要促進(jìn)提高我國(guó)研發(fā)能力以外,與國(guó)際靠攏的審評(píng)政策調(diào)整也有利于為我國(guó)醫(yī)療器械走出國(guó)門做鋪墊����,以引導(dǎo)我國(guó)產(chǎn)品與國(guó)際標(biāo)準(zhǔn)接軌。
3.4 完善專利制度��,樹立可信可靠國(guó)家形象
實(shí)現(xiàn)進(jìn)口創(chuàng)新器械在境內(nèi)外的同步上市,需要在產(chǎn)品研發(fā)早期就在我國(guó)申報(bào)專利��,向?qū)徳u(píng)機(jī)構(gòu)遞交未公開的研發(fā)細(xì)節(jié)文件,并將尚未完全曝光的樣品送到相關(guān)部門檢測(cè)�����,這些都需要我國(guó)在國(guó)際上建立可信可靠的形象�����,取得進(jìn)口企業(yè)的信任�����,具體措施包括:完善專利及知識(shí)產(chǎn)權(quán)管理保護(hù)工作����,將專利保護(hù)制度落實(shí)到研發(fā)�����、檢測(cè)�����、臨床��、上市審批及上市后監(jiān)管全過程��,充分保護(hù)創(chuàng)新成果����。同時(shí)鼓勵(lì)我國(guó)創(chuàng)新醫(yī)療器械企業(yè)盡早進(jìn)行全球?qū)@季?����,為日后產(chǎn)品走出國(guó)門做準(zhǔn)備��。
3.5 允許創(chuàng)新產(chǎn)品自主靈活定價(jià)��,為全球統(tǒng)一定價(jià)提供空間
2022 年9 月�����, 在《國(guó)家醫(yī)療保障局對(duì)十三屆全國(guó)人大五次會(huì)議第4955 號(hào)建議的答復(fù)》中�����,答復(fù)了關(guān)于將創(chuàng)新產(chǎn)品納入醫(yī)保綠色支付通道的建議����,并指出:由于創(chuàng)新醫(yī)療器械臨床使用尚未成熟、使用量暫時(shí)難以預(yù)估����,尚難以實(shí)施帶量方式。在集中帶量采購(gòu)之外留出一定市場(chǎng)為創(chuàng)新產(chǎn)品開拓市場(chǎng)提供空間[30]�����。無(wú)論是國(guó)產(chǎn)還是進(jìn)口企業(yè)��,創(chuàng)新醫(yī)療器械都面臨著研發(fā)周期長(zhǎng)、投入大�����、醫(yī)師培訓(xùn)任務(wù)重����、市場(chǎng)推廣難以及回收成本時(shí)間漫長(zhǎng)的問題��,暫時(shí)不納入集采有利于給創(chuàng)新企業(yè)留出合理的利潤(rùn)空間��,發(fā)揮市場(chǎng)經(jīng)濟(jì)的作用促進(jìn)發(fā)展,對(duì)于進(jìn)口企業(yè)也可以更靈活地實(shí)現(xiàn)全球統(tǒng)一定價(jià)管理。
3.6 鼓勵(lì)境內(nèi)企業(yè)以多種形式開展海外合作并以國(guó)產(chǎn)申請(qǐng)人身份注冊(cè)申報(bào)
根據(jù)《關(guān)于公布醫(yī)療器械注冊(cè)申報(bào)資料要求和批準(zhǔn)證明文件格式的公告》附件8 中的要求����,境內(nèi)申請(qǐng)人應(yīng)提交企業(yè)營(yíng)業(yè)執(zhí)照或事業(yè)單位法人證書��,而境外申請(qǐng)人應(yīng)提交企業(yè)資質(zhì)證明文件及境外上市批件或創(chuàng)新批件。由于臨床試驗(yàn)審批中對(duì)進(jìn)口醫(yī)療器械和國(guó)產(chǎn)醫(yī)療器械的要求不同��,將可能影響進(jìn)口產(chǎn)品的全球同步研發(fā)��,難以把我國(guó)列入國(guó)際多中心臨床試驗(yàn)�����。但若進(jìn)口企業(yè)與我國(guó)相關(guān)企業(yè)進(jìn)行合作并以境內(nèi)申請(qǐng)人的身份申報(bào),則可以更早地進(jìn)入臨床階段�����。因此��,以多種方式參與海外創(chuàng)新活動(dòng)也是促進(jìn)國(guó)際技術(shù)交流��、提升我國(guó)研發(fā)能力��、帶動(dòng)上下游產(chǎn)業(yè)發(fā)展的重要途徑����,包括聯(lián)合開發(fā)����、合并收購(gòu)以及委托生產(chǎn)等�����。例如����,沛嘉醫(yī)療科技(蘇州)有限公司與法國(guó)HighLifeSAS 公司簽署了合作協(xié)議����,獲得了法國(guó)HighLife SAS 公司經(jīng)導(dǎo)管二尖瓣置換術(shù)有關(guān)產(chǎn)品的獨(dú)家許可����,將負(fù)責(zé)該產(chǎn)品在我國(guó)的制造��、開發(fā)和商業(yè)化�����。作為需要臨床試驗(yàn)審批的第三類高風(fēng)險(xiǎn)產(chǎn)品,沛嘉醫(yī)療科技(蘇州)有限公司利用這種合作模式將進(jìn)口技術(shù)以境內(nèi)申請(qǐng)人的身份引入我國(guó)�����,加快了該產(chǎn)品的上市進(jìn)程�����。
4、結(jié)語(yǔ)
自創(chuàng)新醫(yī)療器械注冊(cè)程序?qū)嵤┮詠?lái)�����,我國(guó)通過該途徑申報(bào)和獲批的產(chǎn)品逐年增加��,我國(guó)各級(jí)部門包括醫(yī)療器械監(jiān)管部門����、檢測(cè)機(jī)構(gòu)����、臨床試驗(yàn)機(jī)構(gòu)均出臺(tái)了相應(yīng)政策�����,縮短了審評(píng)審批時(shí)長(zhǎng),推動(dòng)創(chuàng)新醫(yī)療器械的加速上市,但尚未達(dá)到與境外同步上市的目標(biāo)����。為進(jìn)一步實(shí)現(xiàn)進(jìn)口創(chuàng)新醫(yī)療器械在我國(guó)同步上市的目標(biāo),本文提出了相關(guān)政策建議,以促進(jìn)進(jìn)口創(chuàng)新醫(yī)療器械在我國(guó)同步上市。相關(guān)建議不僅使進(jìn)口企業(yè)受益,吸引全球資本����,也有利于促進(jìn)我國(guó)企業(yè)與國(guó)際標(biāo)準(zhǔn)接軌,提升專利布局意識(shí)��,加強(qiáng)國(guó)際合作�����,不斷縮短與國(guó)際先進(jìn)水平的差距��,實(shí)現(xiàn)以人民健康為中心的發(fā)展目標(biāo)����。
參考文獻(xiàn)
[1] 國(guó)家發(fā)展和改革委員會(huì). 中華人民共和國(guó)國(guó)民經(jīng)濟(jì)和社會(huì)發(fā)展第十四個(gè)五年規(guī)劃和2035 年遠(yuǎn)景目標(biāo)綱要[EB/OL]. (2021-03-13) .https://
www.gov.cn/xinwen/2021-03/13/content_5592681.htm#:~:text=%E4%B8%AD%E5%8D%8E%E4%BA%BA%E6%B0%91%E5%85%B1%E5%92%8C%E5%9B%BD%E5%9B%BD
,%E5%85%B1%E5%90%8C%E7%9A%84%E8%A1%8C%E5%8A%A8%E7%BA%B2%E9%A2%86%E3%80%82.
[2] 工業(yè)和信息化部 國(guó)家發(fā)展和改革委員會(huì) 科學(xué)技術(shù)部 商務(wù)部 國(guó)家衛(wèi)生健康委員會(huì) 應(yīng)急管理部 國(guó)家醫(yī)療保障局 國(guó)家藥品監(jiān)督管理局
國(guó)家中醫(yī)藥管理局關(guān)于印發(fā)“十四五”醫(yī)藥工業(yè)發(fā)展規(guī)劃的通知 [EB/OL]. (2022-01-31) . https://www.gov.cn/zhengce/zhengceku/2022-01/31/content_5671480.htm.
[3] 國(guó)家藥品監(jiān)督管理局��,國(guó)家發(fā)展和改革委員會(huì)�����,科學(xué)技術(shù)部����,等.“十四五”國(guó)家藥品安全及促進(jìn)高質(zhì)量發(fā)展規(guī)劃 [EB/OL].(2021-12-30).
https://www.nmpa.gov.cn/directory/web/nmpa///yaopin/ypjgdt/20211230145247117.html.
[4] 國(guó)家市場(chǎng)監(jiān)督管理總局. 醫(yī)療器械注冊(cè)與備案管理辦法[EB/OL].(2021-08-26) .https://www.samr.gov.cn/zw/zfxxgk/fdzdgknr/fgs/art/2023/art_568880e3ee344c45b38d073bba1c53ad.html.
[5] 國(guó)務(wù)院. 醫(yī)療器械監(jiān)督管理?xiàng)l例[EB/OL]. (2021-03-19). https://www.gov.cn/zhengce/content/2021-03/18/content_5593739.htm.
[6] 王蘭明, 趙陽(yáng). 深化醫(yī)療器械審評(píng)審批制度改革, 促進(jìn)醫(yī)療器械產(chǎn)業(yè)高質(zhì)量發(fā)展:中國(guó)醫(yī)療器械審評(píng)審批制度改革概述[J]. 中國(guó)食品藥品監(jiān)管,2021(1):16-28.
[7] 國(guó)家食品藥品監(jiān)督管理總局. 關(guān)于印發(fā)創(chuàng)新醫(yī)療器械特別審批程序( 試行) 的通知[EB/OL].(2014-02-07) .https://www.nmpa.gov.cn/directory/web/nmpa/xxgk/fgwj/gzwj/gzwjylqx/20140207154501788.html.
[8] 國(guó)家藥品監(jiān)督管理局. 關(guān)于發(fā)布創(chuàng)新醫(yī)療器械特別審查申報(bào)資料編寫指南的通告(2018 年第127 號(hào))[EB/OL].(2018-12-12) .https://www.nmpa.gov.cn/xxgk/ggtg/ylqxggtg/ylqxqtggtg/20181105160001106.html.
[9] 國(guó)家藥品監(jiān)督管理局醫(yī)療器械技術(shù)審評(píng)中心. 關(guān)于發(fā)布醫(yī)療器械技術(shù)審評(píng)中心創(chuàng)新醫(yī)療器械特別審查申請(qǐng)審查操作規(guī)范的通告(2018 年第11 號(hào))[EB/OL]. (2018-11-29).https://www.cmde.org.cn//xwdt/shpgzgg/gztg/20181129151100780.html.
[10] FDA. Breakthrough Devices Program[EB/OL]. (2023-02-24).https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program.
[11] FDA. Safer Technologies Program (SteP) for Medical Devices[EB/OL]. (2021-03-18).https://www.fda.gov/medical-devices/how-study-andmarket-your-device/safer-technologies-program-step-medical-devices.
[12] 國(guó)家發(fā)展改革委,國(guó)家衛(wèi)生健康委����,國(guó)家中醫(yī)藥局,等. 關(guān)于支持建設(shè)博鰲樂城國(guó)際醫(yī)療旅游先行區(qū)的實(shí)施方案[EB/OL].(2019-09-10).
https://www.gov.cn/xinwen/2019-09/17/content_5430452.htm.
[13] FDA. Investigational Device Exemption (IDE)[EB/OL]. (2022-10-03). https://www.fda.gov/medical-devices/premarket-submissionsselecting-and-preparing-correct-submission/investigational-device-exemption-ide.
[14] 國(guó)家藥品監(jiān)督管理局. 國(guó)家藥監(jiān)局關(guān)于發(fā)布需進(jìn)行臨床試驗(yàn)審批的第三類醫(yī)療器械目錄(2020 年修訂版)的通告(2020 年第61 號(hào))[EB/OL].(2020-09-18). https://www.nmpa.gov.cn/xxgk/ggtg/ylqxggtg/ylqxqtggtg/20200918103742111.html.
[15] The Federal Register. 42 CFR Part 11 [EB/OL].(2016-09-21). https://www.ecfr.gov/current/title-42/chapter-I/subchapter-A/part-11.
[16] FDA. Changes or Modifications During the Conduct of a Clinical Investigation; Final Guidance for Industry and CDRH Staff [EB/OL].(2001-05-29). https://www.fda.gov/media/72429/download.
[17] The Federal Register. 21 CFR 820. Quality System Regulation [EB/OL]. (2023-03-01). https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820.
[18] The Federal Register. 21 CFR 58 Good Laboratory Practice for Nonclinical Laboratory Studies [EB/OL]. (2023-12-22). https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-58.
[19] 國(guó)家藥品監(jiān)督管理局. 國(guó)家藥監(jiān)局關(guān)于發(fā)布《醫(yī)療器械注冊(cè)自檢管理規(guī)定》的公告(2021 年第126 號(hào))[EB/OL]. (2021-10-22).https://www.nmpa.gov.cn/xxgk/fgwj/xzhgfxwj/20211022153823130.html.
[20] 王保亭����,耿鴻武. 醫(yī)療器械藍(lán)皮書:中國(guó)醫(yī)療器械行業(yè)發(fā)展報(bào)告(2022)[M]. 北京: 社會(huì)科學(xué)文獻(xiàn)出版社,2022.
[21] FDA. FDA and Industry Actions on Premarket Notification (510(k)) Submissions: Effect on FDA Review Clock and Goals [EB/OL]. (2022-10-03).www.fda.gov/media/73507/download.
[22] FDA. FDA and Industry Actions on Premarket Approval Applications (PMAs): Effect on FDA Review Clock and Goals [EB/OL]. (2022-10-03).
www.fda.gov/media/73504/download.
[23] FDA. FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review Clock and Goals [EB/OL]. (2022-10-03).www.fda.gov/media/107652/download.
[24] 國(guó)家藥品監(jiān)督管理局. 關(guān)于公布醫(yī)療器械注冊(cè)申報(bào)資料要求和批準(zhǔn)證明文件格式的公告(2021 年第121 號(hào))[EB/OL].(2021-09-30).
https://www.nmpa.gov.cn/ylqx/ylqxggtg/20210930155134148.html.
[25] 國(guó)家食品藥品監(jiān)督管理總局. 總局關(guān)于發(fā)布接受醫(yī)療器械境外臨床試驗(yàn)數(shù)據(jù)技術(shù)指導(dǎo)原則的通告(2018 年第13 號(hào))[EB/OL].(2018-01-11).
https://www.nmpa.gov.cn/xxgk/ggtg/ylqxggtg/ylqxqtggtg/20180111175801772.html.
[26] 國(guó)家藥品監(jiān)督管理局. 關(guān)于發(fā)布真實(shí)世界數(shù)據(jù)用于醫(yī)療器械臨床評(píng)價(jià)技術(shù)指導(dǎo)原則(試行)的通告[EB/OL].(2020-11-26) .https://www.nmpa.gov.cn/xxgk/ggtg/ylqxggtg/ylqxqtggtg/20201126090030150.html.
[27] 國(guó)家藥品監(jiān)督管理局藥品審評(píng)中心. 國(guó)家藥監(jiān)局藥審中心關(guān)于發(fā)布《藥物臨床試驗(yàn)期間方案變更技術(shù)指導(dǎo)原則(試行)》的通告(2022 年第34 號(hào))[EB/OL].(2022-06-23). https://www.cde.org.cn/main/news/viewInfoCommon/c9d649a44ba90b52ceb8072c28da768f.
[28] 國(guó)家藥品監(jiān)督管理局�����,國(guó)家標(biāo)準(zhǔn)化管理委員會(huì). 關(guān)于進(jìn)一步促進(jìn)醫(yī)療器械標(biāo)準(zhǔn)化工作高質(zhì)量發(fā)展的意見[EB/OL]. (2021-03-30). https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjylqx/20210330170905141.html.
[29] 許慧雯, 孟蕓, 邵姝姝, 等. 監(jiān)管法規(guī)協(xié)調(diào)背景下醫(yī)療器械國(guó)際標(biāo)準(zhǔn)轉(zhuǎn)化研究與思考[J]. 中國(guó)藥事,2022,36(12):1350-1357.