藥物研發(fā)中雜質(zhì)的研究貫穿其整個(gè)階段����,是保證藥品質(zhì)量的重要內(nèi)容。藥品中的雜質(zhì)一般分為三類:有機(jī)雜質(zhì)�、無(wú)機(jī)雜質(zhì)及殘留溶劑。
有機(jī)雜質(zhì)是指在藥品的生產(chǎn)與儲(chǔ)存過(guò)程中引入的有機(jī)化合物雜質(zhì)�,這些雜質(zhì)可以是已知的、未知的����、揮發(fā)性的或不揮發(fā)性的雜質(zhì)���。其來(lái)源多種多樣,主要包括:降解產(chǎn)物����、聚合物、原料藥與輔料或內(nèi)包材的反應(yīng)產(chǎn)物���、以及原料藥制備過(guò)程中引入的起始原料���、副產(chǎn)物、中間體���、反應(yīng)試劑、配位體與催化劑����。
無(wú)機(jī)雜質(zhì)是指在藥品的生產(chǎn)過(guò)程中引入的無(wú)機(jī)物質(zhì),這些雜質(zhì)通常是已知的�,主要包括:反應(yīng)試劑、配位體與催化劑����、重金屬或其它殘留的金屬����、無(wú)機(jī)鹽�、過(guò)濾助劑、活性炭等其它物質(zhì)�。
藥品中的殘留溶劑系指原料藥或輔料的生產(chǎn)中,以及制劑制備過(guò)程中使用的����,但在工藝操作過(guò)程中未能完全去除的有機(jī)溶劑,一般具有已知的毒性����。
雜質(zhì)研究及控制是藥品安全保證的關(guān)鍵要素之一,是藥品研發(fā)中風(fēng)險(xiǎn)控制意識(shí)的重要體現(xiàn),藥品臨床使用中的不良反應(yīng)除了與藥品本身的藥理活性有關(guān)外����,有時(shí)還與藥品中的雜質(zhì)有關(guān),須嚴(yán)格控制����。
ICH Q3藥物雜質(zhì)指導(dǎo)原則
藥物原料或制劑中的雜質(zhì)可能引起臨床不良反應(yīng)。雜質(zhì)毒理學(xué)評(píng)估是藥物研究的重要內(nèi)容����?���;瘜W(xué)合成藥的原藥和制劑中雜質(zhì)的毒性���,尤其是遺傳毒性�,是影響藥物安全性的重要因素����。
ICH (International Council on Harmonization of Technical Requirements for Pharmaceuticals for Human Use, 國(guó)際人用藥品注冊(cè)技術(shù)要求協(xié)調(diào)會(huì))制定了如何控制藥品雜質(zhì)的指導(dǎo)原則,現(xiàn)行指導(dǎo)原則包括:
Q3A (R2):新原料藥中的雜質(zhì)(Impurities in New Drug Substances)
Q3A主要闡述了原料藥中對(duì)雜質(zhì)控制的整體思路���、方法和限度����。Q3A中規(guī)定了新活性藥物成分(active pharmaceutical ingredient, api)中雜質(zhì)需要進(jìn)行報(bào)告����、鑒定和界定的限度����。如雜質(zhì)含量超過(guò)鑒定限度或達(dá)到質(zhì)控限度,藥物研發(fā)者有義務(wù)對(duì)雜質(zhì)的安全性進(jìn)行評(píng)價(jià)�。
Q3B (R2):新藥制劑中的雜質(zhì)(Impurities in New Drug Products)
Q3B闡述了新藥制劑中雜質(zhì)控制����,包括藥物原料的降解產(chǎn)物或者藥物與賦形劑和/或直接接觸容器的反應(yīng)產(chǎn)物���。Q3B中規(guī)定了新藥制劑中降解產(chǎn)物的報(bào)告�、鑒定和界定的限度�。
Q3C (R8):雜質(zhì):殘留溶劑的指導(dǎo)原則(Impurities:Guideline for Residual Solvents)
Q3C引入了每日最大允許量(permitted daily exposure, pde)的概念,指藥物中的雜質(zhì)每日最大可接受而不產(chǎn)生毒性的攝入量�,同時(shí)描述了PDE的計(jì)算方式。Q3C依據(jù)可能的健康風(fēng)險(xiǎn)程度�,將常見(jiàn)的藥物制劑中殘留溶劑分為3類,并規(guī)定了相應(yīng)殘留溶劑的PDE值�,如二甲苯的PDE值為21.7 mg/天。
Q3D (R2):元素雜質(zhì)指導(dǎo)原則(Guideline for Elemental Impurities)
Q3D對(duì)元素雜質(zhì)的控制思路和策略類似于殘留溶劑的控制�。根據(jù)元素雜質(zhì)的毒性和制劑中存在的可能性,Q3D將24種元素雜質(zhì)分為四類���,并給出不同暴露途徑下相應(yīng)PDE值�。
毒理學(xué)風(fēng)險(xiǎn)評(píng)估
傳統(tǒng)的毒理學(xué)評(píng)估主要是基于動(dòng)物試驗(yàn)����,但體內(nèi)試驗(yàn)復(fù)雜且費(fèi)用高昂,同時(shí)由于保障動(dòng)物福利等原因�,故在化學(xué)品安全評(píng)估中����,需要優(yōu)先考慮減少動(dòng)物測(cè)試�,要求更積極地采用動(dòng)物替代方法來(lái)獲取化合物特性數(shù)據(jù)?;衔锏亩纠韺W(xué)風(fēng)險(xiǎn)評(píng)估是利用相關(guān)數(shù)據(jù)庫(kù)和文獻(xiàn)資料,針對(duì)特定化合物進(jìn)行風(fēng)險(xiǎn)評(píng)估的方法�。其檢索評(píng)估的成本遠(yuǎn)低于生物學(xué)試驗(yàn)。因此結(jié)合文獻(xiàn)資料和數(shù)據(jù)庫(kù)進(jìn)行毒理學(xué)風(fēng)險(xiǎn)評(píng)估已逐步代替生物學(xué)試驗(yàn)����。部分監(jiān)管單位如美國(guó)FDA也建議結(jié)合化合物的毒理學(xué)數(shù)據(jù)來(lái)開展風(fēng)險(xiǎn)評(píng)估。
1.毒理學(xué)評(píng)估流程
毒理學(xué)數(shù)據(jù)來(lái)源:
權(quán)威機(jī)構(gòu)文件:ICH����、FDA、U.S.EPA�、WHO/JECFA、EFSA�、OECD和ATSDR等
公開數(shù)據(jù)庫(kù):ECHA、HSDB等
2.非基因毒雜質(zhì)的評(píng)估(PDE法)
對(duì)于非基因毒性化合物或具有實(shí)際閾值的基因毒物質(zhì)����,根據(jù)ICH Q3C和ICH Q3D指導(dǎo)原則推導(dǎo)PDE值�。
PDE值是由POD(Point of Departure)結(jié)合不確定因子推導(dǎo)得到����,PDE的計(jì)算公式如下:
PDE = POD × BW ÷ (F1 × F2 × F3 × F4 × F5 ×F6)
POD是檢索獲得的人類或動(dòng)物相關(guān)毒理學(xué)試驗(yàn)的數(shù)據(jù)�,是在對(duì)化合物的全身毒性���、基因毒性���、致癌性���、生殖發(fā)育毒性����、神經(jīng)毒性等生物學(xué)終點(diǎn)進(jìn)行全面考察�,并依據(jù)產(chǎn)品的接觸途徑、接觸人群�、接觸時(shí)間等信息來(lái)綜合判斷選定的,如NOAEL (No observed adverse effect level)����,LOAEL (Lowest observed adverse effect level),BMDL (Benchmark dose low)�,RfD (Reference Dose)等。
BW為質(zhì)量調(diào)整系數(shù),ICH假設(shè)任何性別的成人體重均為50 kg����。
F1~F6:不確定因子,對(duì)選擇的POD進(jìn)行種內(nèi)差異�、種間差異、暴露時(shí)間�、途徑轉(zhuǎn)換等修正。
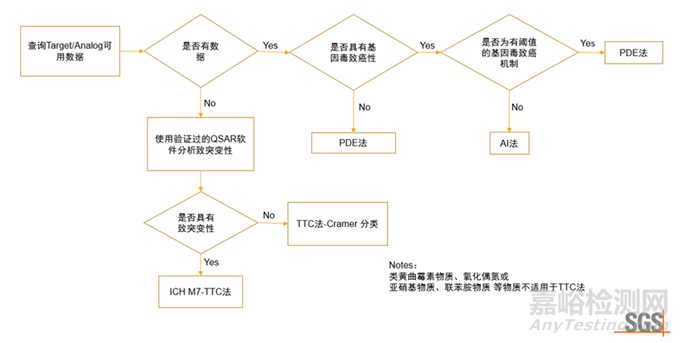
3.基因毒雜質(zhì)的評(píng)估
基因毒性雜質(zhì)(genotoxic impurity����,GTI)是指藥物中能直接或間接導(dǎo)致DNA受損引起基因突變,并具有致癌性或者潛在致癌可能性的一類雜質(zhì)���?��;蚨拘噪s質(zhì)是受到藥品監(jiān)管機(jī)構(gòu)和制藥企業(yè)重點(diǎn)關(guān)注和控制的對(duì)象,由于其較一般雜質(zhì)具有微量水平就存在潛在致突變性和致癌性風(fēng)險(xiǎn)的特點(diǎn)�,需要嚴(yán)格控制其在藥物中的含量以保證藥物質(zhì)量與臨床應(yīng)用的安全性。
藥物雜質(zhì)研究中���,首先基于警示結(jié)構(gòu)�,鑒別并分離出其中可能的基因毒性雜質(zhì)�。通過(guò)數(shù)據(jù)庫(kù)文獻(xiàn)檢索�、定量結(jié)構(gòu)-效應(yīng)關(guān)系(Quantitative Structure-Activity Relationship, QSAR)評(píng)估和體內(nèi)外相關(guān)毒理學(xué)試驗(yàn)�,對(duì)可能存在的基因毒性雜質(zhì)進(jìn)行分類并確定可接受的限度標(biāo)準(zhǔn)。
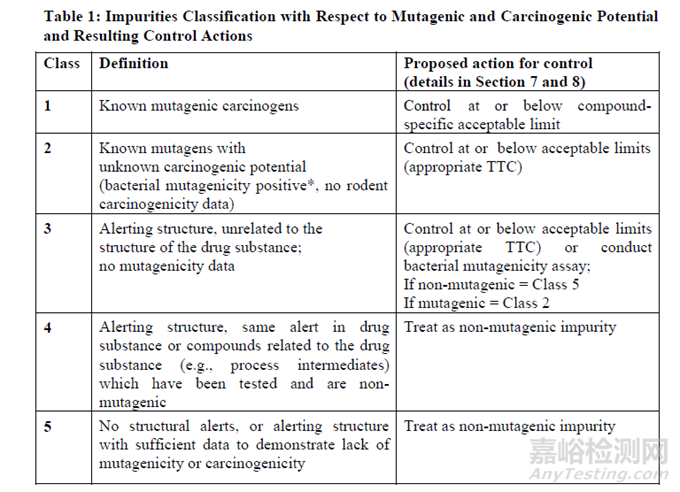
ICH M7 (R2) 評(píng)估和控制藥物中DNA反應(yīng)性(致突變)雜質(zhì)以限值潛在致癌風(fēng)險(xiǎn)(Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk) 根據(jù)雜質(zhì)的致突變性和致癌性分為5類����。
3.1.有數(shù)據(jù)物質(zhì)的限值推導(dǎo)(AI法)
對(duì)于已知的基因毒性致癌類化合物�,可以根據(jù)其致癌性數(shù)據(jù)(TD50或SF)和線性外推的方法計(jì)算出該化合物特定的AI(acceptable intake)值。具體有以下兩種方法�。
TD50法:當(dāng)動(dòng)物致癌試驗(yàn)的數(shù)據(jù)可用時(shí),將TD50(腫瘤發(fā)生率為50%時(shí)對(duì)應(yīng)的劑量����,相當(dāng)于致癌風(fēng)險(xiǎn)水平為1:2)線性外推至致癌風(fēng)險(xiǎn)為10-5時(shí)對(duì)應(yīng)的劑量,以此得到AI值���。
AI(mg/kg/天)= TD50(mg/kg/天)÷ 50,000
SF法:斜率因子(slope factor)是暴露于1 mg/kg/天致癌化合物的單位癌癥發(fā)生率���。當(dāng)斜率因子可用時(shí),可以通過(guò)計(jì)算終生致癌風(fēng)險(xiǎn)為10-5時(shí)對(duì)應(yīng)的劑量得到AI值����。
AI(mg/kg/天)= 10-5 ÷ SF(mg/kg/天)-1
對(duì)于有數(shù)據(jù)支持為非基因毒性化合物或具有實(shí)際閾值的基因毒物質(zhì),根據(jù)ICH Q3C和ICH Q3D指導(dǎo)原則推導(dǎo)PDE值���。計(jì)算方法同上述非基因毒物質(zhì)PDE推導(dǎo)方法�。使用該方法時(shí)需要更多的毒理學(xué)數(shù)據(jù),證明閾值機(jī)制的存在�。
3.2.無(wú)數(shù)據(jù)物質(zhì)的限值確定(QSAR軟件預(yù)測(cè)和TTC法)
根據(jù)ICH M7中的描述,危害性評(píng)估首先通過(guò)對(duì)數(shù)據(jù)庫(kù)和文獻(xiàn)的檢索獲得雜質(zhì)致癌性和細(xì)菌致突變數(shù)據(jù)對(duì)實(shí)際和潛在雜質(zhì)進(jìn)行初步分析�,并將其歸為1、2或5類�。如果無(wú)法獲得化合物的致突變和致癌性數(shù)據(jù)及分類依據(jù),則可進(jìn)行定量結(jié)構(gòu)-效應(yīng)關(guān)系(QSAR)預(yù)測(cè)�,如預(yù)測(cè)細(xì)菌回復(fù)突變性。常見(jiàn)的QSAR模型可分為專家知識(shí)模型(rule-based model)和統(tǒng)計(jì)學(xué)模型(statistic-based model)����。專家知識(shí)模型是基于專家經(jīng)驗(yàn)制定的各種規(guī)則所建立的;而統(tǒng)計(jì)學(xué)模型則是基于回歸分析�、偏最小二乘、神經(jīng)網(wǎng)絡(luò)等數(shù)學(xué)模型所建立�。如果經(jīng)兩種互補(bǔ)的QSAR方法(專家知識(shí)模型和統(tǒng)計(jì)學(xué)模型)預(yù)測(cè)均無(wú)致突變性,則可認(rèn)為該雜質(zhì)沒(méi)有致突變性�,不建議做進(jìn)一步的檢測(cè),可以判定該雜質(zhì)為5類���。
當(dāng)缺乏足夠的毒理學(xué)數(shù)據(jù)推導(dǎo)化合物的AI值����,且QSAR預(yù)測(cè)細(xì)菌回復(fù)突變?yōu)殛?yáng)性時(shí),應(yīng)使用M7 TTC法���。ICH M7中的毒理學(xué)關(guān)注閾值(Threshold of Toxicological Concern���,TTC),低于該水平時(shí)�,可以認(rèn)為暴露于該化合物對(duì)人類健康無(wú)明顯風(fēng)險(xiǎn)����。在使用TTC評(píng)估原料藥和制劑中致突變雜質(zhì)的可接受限度時(shí),終生限度為1.5 μg/天����。若存在同類基因毒雜質(zhì),可根據(jù)多個(gè)雜質(zhì)的可接受攝入量來(lái)制定限度����。
對(duì)于短于終生給藥的藥品中致突變雜質(zhì)的TTC值可調(diào)整為更高劑量,不同給藥時(shí)長(zhǎng)的單個(gè)雜質(zhì)TTC如下:
|
治療期 |
≤1月 |
>1月-12月 |
>1-10年 |
>10年 |
|
TTC(μg/天) |
120 |
20 |
10 |
1.5 |
3.3. 遺傳毒性試驗(yàn)
當(dāng)QSAR軟件評(píng)估致突變得到陽(yáng)性結(jié)果時(shí)�,也可進(jìn)一步進(jìn)行體外評(píng)估(如:Ames試驗(yàn))。如果Ames試驗(yàn)為陰性���,則該雜質(zhì)應(yīng)歸為5類�。如果Ames試驗(yàn)結(jié)果為陽(yáng)性,后續(xù)應(yīng)進(jìn)行體內(nèi)試驗(yàn)����,明確體內(nèi)致突變風(fēng)險(xiǎn)。
小結(jié):
PDE法多用于非基因毒致癌物�。對(duì)于致癌性數(shù)據(jù)為陽(yáng)性的致突變雜質(zhì),需使用AI法進(jìn)行評(píng)估���。在目標(biāo)化合物數(shù)據(jù)無(wú)法獲得的情況下���,可以通過(guò)QSAR預(yù)測(cè)并使用TTC值進(jìn)行評(píng)估。
毒理學(xué)風(fēng)險(xiǎn)評(píng)估的應(yīng)用
1.藥包材相容性
藥物在儲(chǔ)存過(guò)程中���,有效成分會(huì)持續(xù)與主要包裝系統(tǒng)的材料發(fā)生相互作用�。包裝材料中的物質(zhì)可能會(huì)遷移到藥物中���,對(duì)藥效產(chǎn)生不利影響或者導(dǎo)致毒性�?���?商崛∥锖涂山鑫?E&Ls, extractable and leachables)研究可提供其相互作用的數(shù)據(jù),從而評(píng)估其潛在風(fēng)險(xiǎn)���。任何測(cè)得超過(guò)分析評(píng)估閾值(AET)的化合物都應(yīng)該被識(shí)別出來(lái)并報(bào)告其檢出量����,該檢出量應(yīng)根據(jù)藥品使用方式轉(zhuǎn)化為患者潛在暴露的估計(jì)值,再與化合物對(duì)應(yīng)的允許限量(如PDE)進(jìn)行比較����,判斷風(fēng)險(xiǎn)。
2.雜質(zhì)限度
當(dāng)企業(yè)在新藥研發(fā)過(guò)程中�,某特定雜質(zhì)超出了ICH Q3的限度要求,應(yīng)當(dāng)除去這個(gè)雜質(zhì)或降低到ICH Q3要求的限度水平����,但這可能是非常困難或代價(jià)非常高的����。那么可以根據(jù)該雜質(zhì)的毒性水平制定其PDE值。PDE與限度本質(zhì)上等同�,都用于表示雜質(zhì)控制的水平,不同在于ICH Q3中的限度通常以百分比(%)表示�,而PDE為某種化合物的特定限度,是根據(jù)其毒性數(shù)據(jù)專門推導(dǎo)得到���,通常用μg/天表示����。PDE除以產(chǎn)品的最大日服用劑量,得出特定化合物的安全限度值���。
3.共線生產(chǎn)中的殘留限度
當(dāng)一個(gè)共用設(shè)施替代多個(gè)專用設(shè)施生產(chǎn)不同藥品時(shí)�,首先要解決如何防止交叉污染的問(wèn)題���。產(chǎn)線上的一種藥品的殘留對(duì)同產(chǎn)線其他藥品使用的患者來(lái)說(shuō)可能屬于污染物�,從而帶來(lái)潛在風(fēng)險(xiǎn)���。因此���,在共線生產(chǎn)過(guò)程中建立科學(xué)的清潔限度尤其重要?;诮】档谋┞断薅龋╤ealth-based exposure limit,HBEL���,如PDE值)常被用于共線生產(chǎn)中用于設(shè)定清潔驗(yàn)證的殘留限度�。
傳統(tǒng)方法:1/1000最低日治療劑量���、10ppm方法等���。統(tǒng)一限度���,但存在風(fēng)險(xiǎn)和局限。
HBEL法:使用PDE���、ADE(acceptable daily exposure)等����,更加科學(xué)合理����。
2014年歐洲藥品管理局(EMA)發(fā)布的共用設(shè)施中生產(chǎn)不同藥品的暴露限度設(shè)定指南《Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities》和2020年世界衛(wèi)生組織(WHO)發(fā)布的清潔驗(yàn)證指南草案《Points to consider when including Health-Based Exposure Limits (HBEL) in cleaning validation》,均提出需要使用以HBEL為基礎(chǔ)的殘留限度�。
2023年����,中國(guó)國(guó)家藥品監(jiān)督管理局藥品審核查驗(yàn)中心(CFDI)發(fā)布《藥品共線生產(chǎn)質(zhì)量風(fēng)險(xiǎn)管理指南》,其中提到“活性物質(zhì)殘留限度標(biāo)準(zhǔn)應(yīng)當(dāng)基于產(chǎn)品毒理試驗(yàn)數(shù)據(jù)或毒理學(xué)文獻(xiàn)資料結(jié)合實(shí)際生產(chǎn)情況建立�。相對(duì)于傳統(tǒng)方法設(shè)定的限度來(lái)說(shuō),基于健康的暴露限度(HBEL)的可接受標(biāo)準(zhǔn)(如PDE值)在評(píng)估清潔殘留數(shù)據(jù)時(shí)更具有科學(xué)性和優(yōu)勢(shì)”����,體現(xiàn)出國(guó)內(nèi)對(duì)于HBEL方法的關(guān)注度也逐漸提高�。