背景
在過去的半個多世紀里�����,小分子細胞毒素藥物廣泛用于治療各種惡性腫瘤�����,在某些臨床條件下�����,改變了其中一些疾病的自然進程�����。但由于它們內在的作用方式�����,容易引起嚴重的靶外不良事件從而妨礙其臨床療效�����,進而導致早期停藥�����、腫瘤復發(fā)或復發(fā)的風險也隨之增加�����。例如普納替尼(ponatinib)�����,在一項名為PACE的研究中�����,28個月的隨訪期顯示�����,普納替尼增加了心臟毒性的風險�����,包括10%的心血管�����、7%的腦血管和7%的周圍不良事件[1]�����。
為了提高對癌癥患者的療效�����,科學家們研發(fā)出一種可以選擇性靶向腫瘤細胞的細胞毒素藥物�����,以期提高化療藥物的有效性并降至最低全身毒性�����。在這些新穎的藥物中�����,結合了細胞毒性藥物的人源化抗體,即抗體偶聯藥物(Antibody-Drug Conjugate, ADC)�����, 躋身于癌癥科學與臨床發(fā)展的行列[2]�����。2000年�����,FDA首次批準了用于治療成人CD33陽性急性髓性白血病的ADC藥物:Mylotarg® (gemtuzumab ozogamicin)[3]�����,標志著癌癥靶向治療藥物ADC時代的開始�����。2021年6月�����,首個國產ADC藥物�����,榮昌生物研發(fā)的維迪西妥單抗獲批上市�����。ADC藥物技術一直在迅速發(fā)展�����,截止目前�����,全球共有15款ADC藥物獲批上市�����,有200多款ADC藥物正在進行臨床試驗�����。大多數ADC藥物已從I期進展到II期�����,部分ADC藥物的III期試驗顯示出積極的結果。這個新興的化療分子藥物被視為最有效的抗癌治療劑之一�����,預計在不久的將來會顯著增加它的市場份額[4]�����。
什么是ADC
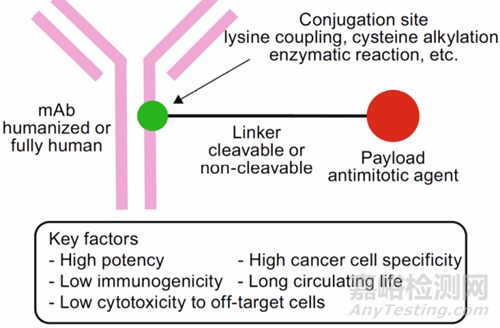
ADC是指通過連接子將小分子化合物偶聯至靶向性抗體或抗體片段的一類生物技術藥物�����,其結構組成包括抗體或抗體片段(Antibody)�����,連接子(Linker)�����,具有細胞毒性的小分子化合物(Payload)(圖1)�����。其中�����,抗體通常選用可被靶細胞內化的抗體�����;連接子具有一定的循環(huán)穩(wěn)定性�����,可以支持藥物在到達靶器官前不被降解或者少量降解�����,進入細胞后能夠快速釋放活性小分子化合物�����;小分子化合物能夠對靶細胞產生藥效作用�����。因此�����,ADC是生物大分子和化學小分子的結合體,具有獨特的作用機制和代謝動力學特征[4]�����。
圖1:ADC結構�����,圖片引自[4]
ADC的作用機制及其優(yōu)勢
ADC藥物中的單克隆抗體表現出選擇性附著和進入抗原過度表達的癌細胞的能力�����。ADC藥物隨后通過一系列細胞內化過程被帶入腫瘤細胞內�����,這一過程從早期含體形成開始�����,發(fā)展到晚期內含體的成熟�����,并最終與溶酶體融合�����。ADC的有效載荷在溶酶體內通過化學或酶作用發(fā)生裂解�����,釋放后�����,這種有效載荷通過破壞目標細胞內的DNA或微管發(fā)揮其細胞毒性�����,最終導致癌細胞的死亡(圖2)�����。此外�����,如果釋放的有效載荷具有透過細胞膜的能力�����,它還能引發(fā)“旁觀者效應”,即不需要ADC的內化作用就能引起鄰近的靶細胞發(fā)生凋亡�����。因此�����,由于ADC藥物精準的抗體結合和有效載荷的促凋亡能力�����,它在癌癥治療方面具有多種優(yōu)勢�����,包括治療窗口及療效增強�����,耐藥性降低�����,癌細胞特異性靶向�����,對正常細胞的毒性相對較低�����,與傳統(tǒng)化學癌癥治療相比�����,副作用較小等[5]�����。
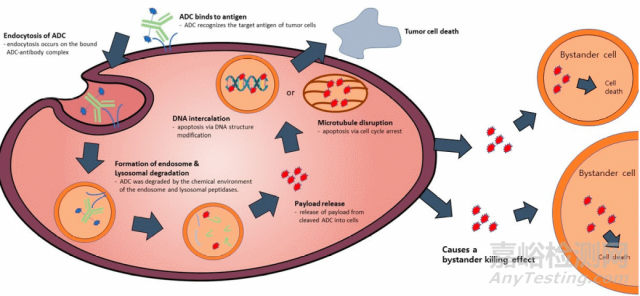
圖2:ADC的作用機制�����,圖片引自[5]
ADC的PK生物分析策略
ADC進入體內后�����,payload可通過酶解或者化學反應從主體結構上逐漸解離�����,主要包括偶聯抗體、抗體偶聯藥物�����、總抗體�����、裸抗�����、游離的小分子payload以及payload相關的代謝產物等�����。他們在體內的濃度變化對解讀ADC的PK藥代動力學特點至關重要�����。ADC為抗體或抗體片段至少結合一個payload(抗體偶聯比(Drug to antibody ratio�����,DAR)≥1);總抗體為未偶聯和偶聯至少一個payload分子的抗體的總和(DAR≥0)�����;Payload為ADC裂解或被分解代謝后形成的游離的payload�����,在FDA于2022年2月發(fā)布的“Clinical Pharmacology Considerations for Antibody-Drug Conjugates”指導原則草案中指出�����,在對ADC藥物進行生物分析時�����,應該測定多種生物分析物用于評估ADC暴露量�����,例如:體內的總抗體含量�����、ADC含量以及小分子payload部分和藥理活性代謝產物的含量[6][7]�����。目前已經上市的ADC藥物的PK檢測主要包括總抗體�����、ADC和小分子化合物部分�����。一般總抗體和ADC的檢測主要在LBA平臺開展�����,小分子化合物部分在LC-MS平臺進行�����。本篇主要對在LBA平臺開展ADC的PK檢測展開討論�����。
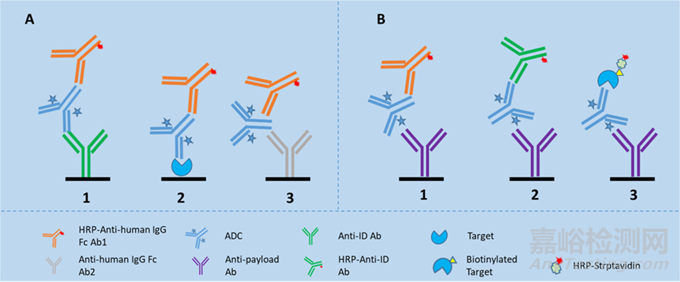
對于總抗體和ADC部分的檢測�����,通常采用ELISA或MSD平臺。針對總抗體�����,通常利用靶點或抗裸抗的特異性抗體作為捕獲試劑進行包被�����,再選擇酶標的抗裸抗特異性抗體或普抗作為檢測抗體進行檢測(圖3A)�����。針對ADC�����,通常利用抗payload特異性抗體作為捕獲抗體進行包被�����,再選擇酶標的抗裸抗特異性抗體�����、普抗或者靶點作為檢測抗體進行檢測(圖3B)�����。以已上市的ADC藥物Enhertu為例�����,它的主體是抗Her2抗體�����,有效載荷為Dxd�����,該上市藥物的PK檢測中�����,總抗體和ADC的檢測是在ECL平臺進行的�����,總抗體的檢測中分別使用anti-HER2 antibody的特異性單克隆抗體作為捕獲試劑和檢測試劑�����,而ADC的檢測中使用抗小分子抗體anti-Dxd antibody作為捕獲抗體,檢測抗體則使用測的是anti-HER2 antibody的單克隆抗體�����。
圖3:總抗體和ADC的PK生物分析�����,圖片引自[8]
ADC的PK生物分析挑戰(zhàn)
由于ADC自身所具有的特殊結構和復雜的組分多樣性�����,其PK生物分析方法的建立具有很大的挑戰(zhàn)性�����,本篇主要針對在LBA開展的ADC的PK檢測展開討論�����,在LBA平臺進行ADC的檢測時主要有以下幾個挑戰(zhàn):(1)DAR值敏感性�����,當ADC進入體內后�����,payload可通過酶解或者化學反應從主體結構上逐漸解離�����,產生不同DAR值的ADC藥物�����,而ADC標準品往往不能準確的反應體內ADC藥物的真實情況�����,尤其是PK靠后的一些采樣時間點樣本[9]�����,另外不同DAR值的ADC藥物與捕獲試劑或者檢測抗體的結合能力可能不同�����,影響總抗體和ADC濃度的準確度測定�����,尤其體現在一些高DAR值的ADC和低DAR值的ADC藥物間。(2)小分子毒素結構的改變�����,目前在研的小分子毒素主要有DNA損傷劑�����、DNA轉錄抑制劑�����、微管蛋白抑制劑�����、美登素�����、喜樹堿類�����、Tubulysin B類似物等�����,其化學結構可能會隨著藥物在體內的生物轉化和分解代謝而發(fā)生改變�����,從而也帶來它獨特的生物分析挑戰(zhàn)�����。例如�����,像喜樹堿類的衍生物�����,作為DNA拓撲異構酶I的抑制劑�����,分子中的內酯環(huán)�����,是主要的活性基團,但結構不穩(wěn)定�����,在pH上升時會打開�����,打開后的結構會改變和抗體之間的結合能力�����,從而影響PK的檢測[10]�����;另外有些小分子毒素在體內會發(fā)生轉化�����,PK的檢測形式也會增多�����,例如ADC藥物MEDI4276�����,臨床的PK檢測包含了總抗體�����,小分子毒素轉化前后的ADC�����,游離的轉化前后的小分子毒素5種形式[11]�����。(3)可溶性靶點蛋白�����,當循環(huán)系統(tǒng)中有高濃度游離的可溶性靶點蛋白時�����,可能會影響抗原或者是特異性抗體和藥物的結合效率�����,從而對PK的檢測帶來一定的影響。(4)抗藥抗體�����,當體內有抗藥抗體或者是中和抗體產生時�����,都會對PK的檢測帶來影響�����,抗藥抗體可能會帶來空間位阻的影響�����,而中和抗體會直接影響抗原或者特異性抗體和藥物的結合�����。
DAR敏感性考量
藥物抗體偶聯比(drug-to-antibody ratio, DAR)是指一個抗體所攜帶payload數量的平均值�����,是ADC最重要的質量屬性之一�����,因為這決定了可以輸送到腫瘤的“有效載荷”�����,可以直接影響安全性和有效性�����。較低的DAR會降低ADC的效力�����,但過高的DAR又會影響ADC分析的藥代動力學和毒性�����。ADC藥物的DAR值通常為0到8之間�����,普遍認為DAR在3-4之間是ADC藥物的最優(yōu)選擇[12]。
ADC進入體內后�����,連接子可能由于酶解或其他的作用發(fā)生斷裂�����,使得payload從主體結構上逐漸解離�����,產生不同形式的ADC�����。以DAR8的ADC為例�����,在進入人體內后可產生DAR6�����、DAR4�����、DAR2等形式的ADC。不同DAR的ADC分子可能會有不同的清除率[12]�����,所以隨著給藥時間的推移�����,不同訪視點采集的樣本中ADC的DAR值分布可能會有很大的不同�����,尤其是在給藥周期比較靠后的時間點�����。因此在建立PK生物分析方法的時候�����,需要考慮檢測方法對不同DAR形式的ADC的敏感性�����,即無論ADC在體內以何種DAR的形式存在�����,都能準確的被檢測到�����。
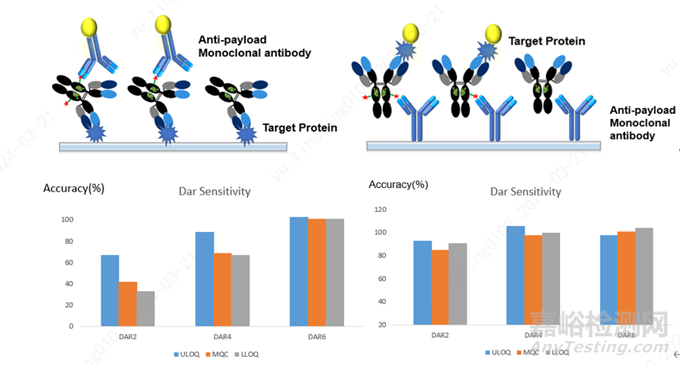
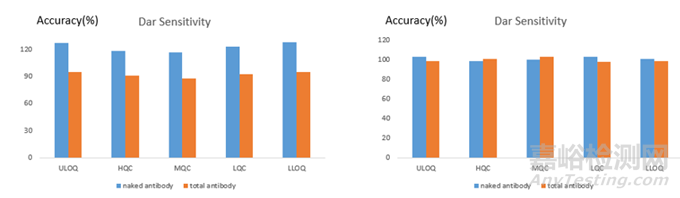
首先對于ADC的檢測�����,可以使用靶點蛋白作為捕獲試劑�����,抗小分子毒素的抗體作為檢測試劑�����,也可以使用抗小分子毒素的抗體作為捕獲試劑�����,使用靶點蛋白作為檢測試劑�����,往往后者在DAR敏感性測試中有比較好的結果,圖4是在一款ADC的PK檢測中使用2種不同assay format在DAR敏感性中的差異�����。從圖中可以看出�����,當使用靶點蛋白作為捕獲試劑�����,抗小分子毒素的抗體作為檢測試劑時�����,隨著DAR值的降低�����,QC的回收率也隨之降低�����;當使用抗小分子毒素的抗體作為捕獲試劑時�����,不同DAR值的ADC間的QC差異較小�����,整體的回收率都在80%-120%之間�����。這和文獻中的報道也比較一致�����,在ADC的檢測中建議使用抗小分子藥物的抗體作為包被試劑�����,不同ADC間檢測的結果更加一致�����。
圖4:ADC檢測中不同assay format下不同DAR值ADC樣品的回收率
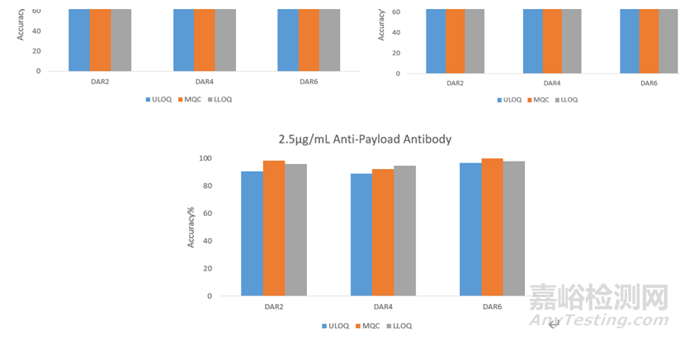
在另一款ADC的檢測中�����,使用抗小分子毒素的抗體作為捕獲試劑,使用抗ADC抗體部分的單克隆抗體作為檢測試劑�����,不同的包被抗體濃度下�����,不同DAR值的ADC樣本間檢測差異也顯著不同�����,見圖5�����,當包被濃度為1.0 ug/ml時�����,DAR=2時的ADC樣本在高濃度�����,中濃度和低濃度質控樣本間的回收率都偏低�����,低于方法中80%-120%的接收標準�����,隨著包被濃度的提高�����,低DAR值的ADC樣本間的回收率逐漸提高�����,當包被濃度提高到2.5 ug/ml時�����,不同DAR值的ADC樣本間檢測的結果較一致�����。
圖5: ADC檢測中不同包被濃度下不同DAR值樣本的回收率
另外在總抗體的檢測中,可以使用靶點蛋白作為捕獲試劑�����,通用性抗體作為檢測試劑�����,但這種assay format情況下�����,可能會出現裸抗的檢測結果遠高于總抗體的情況�����,如圖6中的案列�����,當使用靶點蛋白作為捕獲試劑時�����,裸抗樣本的回收率整體遠高于總抗體�����,并且回收率大多超出方法規(guī)定的接收標準(圖6左)�����,當將靶點蛋白調整為特異性更好的抗體時�����,總抗體和裸抗的檢測結果比較一致(圖6右)�����。
圖6: 總抗體檢測中不同包被試劑在總抗體和裸抗之間的差異
結語
ADC藥物由于自身的特殊結構和復雜的組分多樣性�����,給PK的生物分析方法建立帶來很大的挑戰(zhàn)�����,開發(fā)一款準確而穩(wěn)定的ADC生物分析方法對藥代動力學的研究至關重要�����,一般總抗體和ADC的檢測在LBA平臺展開,小分子化合物的檢測在LC-MS平臺開展�����。在LBA平臺開發(fā)ADC的PK檢測一般需要考慮DAR值敏感性�����、靶點干擾�����、小分子毒素帶來的影響之外�����,也需要關注總抗體和ADC檢測濃度之間的關系�����。
參考文獻
[1] Pun, Shawn C., and Tomas G. Neilan. "Cardiovascular side effects of small molecule therapies for cancer." European Heart Journal 37.36 (2016): 2742-2745.
[2] Hervé Bouchard, Christian Viskov, Carlos Garcia-Echeverria: Antibody–drug conjugates—A new wave of cancer drugs. [J]Bioorganic & Medicinal Chemistry Letters 24 (2014) 5357–5363
[3] Zhiwen Fu, Shijun Li, Sifei Han, Chen Shi1 and Yu Zhang, Antibody drug conjugate: the “biological missile” for targeted cancer therapy, [J]Signal Transduction and Targeted Therapy (2022) 7:93
[4] Kyoji Tsuchikama , Zhiqiang An, Antibody-drug conjugates: recent advances in conjugation and linker chemistries, [J] Protein Cell 2018, 9(1):33–46
[5] Chi Hun Song, Minchan Jeong, Hyukmin In, Ji Hoe Kim, Chih-Wei Lin and Kyung Ho Han, Trends in the Development of Antibody-Drug Conjugates for Cancer Therapy, [J]Antibodies 2023, 12, 72.
[6] Gorovits B, Alley SC, Bilic S, Booth B, Kaur S, Oldfield P, Purushothama S, Rao C, Shord S, Siguenza P. Bioanalysis of antibody-drug conjugates: American Association of Pharmaceutical Scientists Antibody-Drug Conjugate Working Group position paper. Bioanalysis. 5(9):997-1006(2013).
[7] Clinical Pharmacology Considerations for Antibody-Drug Conjugates, Retrieved February 11, 2022.
[8] Qiuping Qin and Likun GongCurrent, Analytical Strategies for Antibody–Drug Conjugates in Biomatrices, [J] Molecules 2022, 27, 6299.
[9] Surinder Kaur, Keyang Xu, Ola M Saad, Randall C Dere & Montserrat Carrasco-Triguero, Bioanalytical assay strategies for the development of antibody–drug conjugate biotherapeutics, Bioanalysis (2013) 5(2), 201–226
[10] Uland Y. Lau, Lauren T. Benoit, Nicole S. Stevens, Kim K. Emmerton, Margo Zaval, Julia H. Cochran,and Peter D. Senter, Lactone Stabilization is Not a Necessary Feature for Antibody Conjugates of Camptothecins, [J]Mol. Pharmaceutics 2018, 15, 4063−4072
[11] Faria, M.; Peay, M.; Lam, B.; Ma, E.; Yuan, M.; Waldron, M.; Mylott Jr, W.R.; Liang, M.; Rosenbaum, A.I. Multiplex LC-MS/MS Assays for Clinical Bioanalysis of MEDI4276, an Antibody-Drug Conjugate of Tubulysin Analogue Attached via Cleavable Linker to a Biparatopic Humanized Antibody against HER-2. Antibodies 2019, 8, 11.
[12] Xiuxia Sun, Jose F. Ponte, Nicholas C. Yoder, Rassol Laleau, Jennifer Coccia, Leanne Lanieri, Qifeng Qiu, Rui Wu, Erica Hong, Megan Bogalhas, Lintao Wang, Ling Dong, Yulius Setiady, Erin K. Maloney, Olga Ab, Xiaoyan Zhang, Jan Pinkas, Thomas A. Keating, Ravi Chari, Hans K. Erickson,§ and John M. Lambert, Effects of Drug−Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody−Maytansinoid Conjugates, Bioconjugate Chem. 2017, 28, 1371−1381