在2022年11月�����,由RDPAC 的藥學(xué)團(tuán)隊(duì)對(duì)中美歐藥學(xué)技術(shù)指導(dǎo)原則和指南進(jìn)行了調(diào)研與對(duì)比�����,總結(jié)分析了中美歐指南的標(biāo)準(zhǔn)差異及實(shí)施情況差異�����,采用了主題詞及分類方式����、深度對(duì)比�、報(bào)告撰寫和定稿流程���,最后呈現(xiàn)研究報(bào)告,給到藥審中心參考�。
2019 年 7 月 16 日,國(guó)家藥監(jiān)局發(fā)布了“關(guān)于進(jìn)一步完善藥品關(guān)聯(lián)審評(píng)審批和監(jiān)管工作事 宜的公告(2019 年第 56 號(hào))�����,該指導(dǎo)原則進(jìn)一步完善了我國(guó)的原輔包與制劑的關(guān)聯(lián)審評(píng)審批 制度��。2020 年 4 月 30 日���,基于新《藥品管理法》和《藥品注冊(cè)管理辦法》發(fā)布的《化學(xué)原料 藥����、藥用輔料及藥包材與藥品制劑關(guān)聯(lián)審評(píng)審批管理規(guī)定(征求意見(jiàn)稿)》�,自發(fā)布以來(lái),仍未落地實(shí)施�。為進(jìn)一步明確關(guān)聯(lián)審評(píng)審批管理要求,解決實(shí)際工作中存在的原輔包登記及生命 周期維護(hù)等問(wèn)題�,建議根據(jù)業(yè)界反饋的意見(jiàn)和建議,盡快落實(shí)該法規(guī)�,便于業(yè)界參考執(zhí)行。推薦加急修訂并發(fā)布����,同時(shí)加強(qiáng)工業(yè)界培訓(xùn)和交流。
國(guó)外相關(guān)參考指南:
21 CFR 314.420 Drug master files - Guideline for Drug Master Files (DMF)
Type II
a) Guidance for Industry M4Q: The CTD - Quality".
b) Guidance for Industry: ANDAs: Stability Testing of Drug Substances and Products: Questions and Answers DRAFT GUIDANCE
c) GDUFA
d) ICH Guideline:Q11 Development and Manufacture of Drug Substances
Type III
a) Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Documentation
b) MAPP 5015.5 CMC Reviews of Type III DMFs for Packaging Materials Type IV
a) DMF Guidance
b) Guidance for Industry: Nonclinical Studies for the Safety Evaluation of Pharmaceutica Excipients
Type V
a) 21 CFR 314.420(a)
Guidance for Industry for the Submission Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products
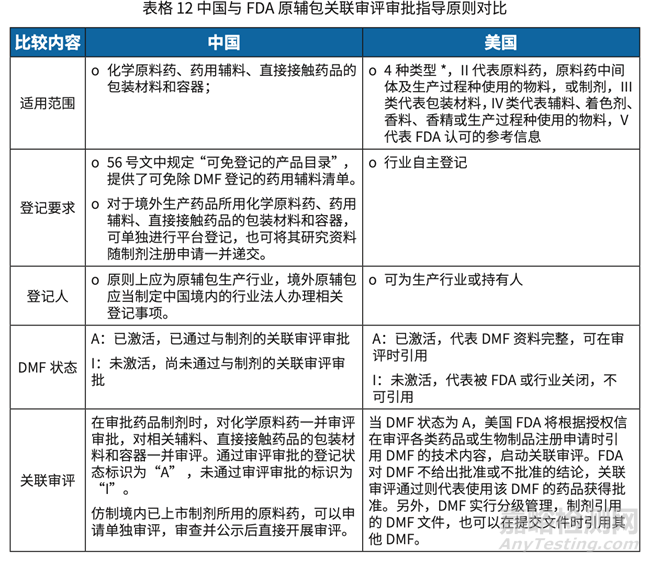

• 中國(guó)實(shí)施實(shí)踐問(wèn)題:
美國(guó) DMF 適用范圍較寬泛���,比如 II 類除了原料藥外���,還包含原料藥中間體以及生產(chǎn)過(guò)程種使用的物料或制劑��。在我國(guó)���,原料藥中間體����、制劑中間體�����,混粉及生物技術(shù)原 料藥等�����,仍未納入登記范圍及關(guān)聯(lián)審評(píng)審批管理�。
在原輔包產(chǎn)品進(jìn)行登記時(shí),同一申請(qǐng)人 / 登記人的多個(gè)生產(chǎn)廠生產(chǎn)的同一產(chǎn)品或同一 產(chǎn)品的不同級(jí)別�,目前不能公用同一個(gè)登記號(hào)。
2020 年 4 月 30 日發(fā)布的關(guān)聯(lián)審評(píng)審批管理規(guī)定����,原輔包行業(yè)授權(quán)使用書模板中要求 提供給藥途徑���,且目前是一個(gè)產(chǎn)品使用一份行業(yè)授權(quán)使用書���。
我國(guó)法規(guī)規(guī)定,原輔包登記人應(yīng)為原輔包生產(chǎn)行業(yè)����,美國(guó)登記人可為生產(chǎn)行業(yè)或持有人, 且允許 DMF 之間進(jìn)行轉(zhuǎn)讓�����。
相較于美國(guó)非活性成分 ( 輔料 ) 數(shù)據(jù)庫(kù),我國(guó)對(duì)于原輔包登記相關(guān)信息公開內(nèi)容無(wú)原 輔包的等級(jí)和用途�����,此信息的公布有利于制劑行業(yè)在選用原輔包時(shí)了解更多信息���。
目前法規(guī)體系中化學(xué)藥品和生物制品的上市后藥學(xué)變更技術(shù)指導(dǎo)原則�,主要針對(duì)已上 市的化學(xué)藥品��,化學(xué)原料藥及生物制品�����,但無(wú)針對(duì)輔料和藥包材相關(guān)技術(shù)指導(dǎo)原則�����, 相關(guān)變更無(wú)參考法規(guī)��。
涉及原輔包來(lái)源變更的補(bǔ)充申請(qǐng)的審評(píng)時(shí)限不明確���。根據(jù) 2020 年 4 月 30 日發(fā)布的關(guān) 聯(lián)審評(píng)審批征求意見(jiàn)稿第 33 條規(guī)定“對(duì)于藥品制劑變更藥用輔料和藥包材來(lái)源的補(bǔ) 充申請(qǐng)���,藥用輔料和藥包材未通過(guò)技術(shù)審評(píng)的,審評(píng)時(shí)限為 130 個(gè)工作日”����,而根據(jù)《藥 品注冊(cè)管理辦法》規(guī)定,上市后補(bǔ)充申請(qǐng)的審評(píng)時(shí)限為 60 個(gè)工作日�。
根據(jù)目前要求和實(shí)際,未登記或登記狀態(tài)為 I 的輔料包材變更�����,制劑上市許可持有人均應(yīng) 通過(guò)補(bǔ)充申請(qǐng)?zhí)峤?����,此類申?qǐng)可能會(huì)觸發(fā)注冊(cè)檢驗(yàn)�。
• 相關(guān)的國(guó)內(nèi)監(jiān)管法規(guī):
1. 2015 年國(guó)務(wù)院發(fā)布 44 號(hào)文�����,嘗試藥用輔料��、藥包材與制劑一并審評(píng)審批�����。
2. 2016 年,總局分別發(fā)布了 134 號(hào)和 155 號(hào)文���,取消了輔料注冊(cè)批件�,啟動(dòng)了關(guān)聯(lián)審評(píng) 審批�,并規(guī)定了輔料包材關(guān)聯(lián)審評(píng)審批資料要求。
3. 2017 年����,總局發(fā)布了 146 號(hào)公告,我國(guó)開始正式實(shí)施原料藥�、藥用輔料和藥包材登與 制劑記和關(guān)聯(lián)審評(píng)政策,并由國(guó)家食品藥品監(jiān)督管理總局藥審中心建立原料藥�����、藥用 輔料和藥包材登記平臺(tái)與數(shù)據(jù)庫(kù)�。
4. 2019 年,國(guó)家藥監(jiān)局發(fā)布了《關(guān)于進(jìn)一步完善藥品關(guān)聯(lián)審評(píng)審批和監(jiān)管工作有關(guān)事宜 的公告》(2019 No. 56)�,規(guī)定原輔包與藥品制劑關(guān)聯(lián)審評(píng)審批由原輔包登記人在登 記平臺(tái)上登記,藥品制劑注冊(cè)申請(qǐng)人提交注冊(cè)申請(qǐng)時(shí)與平臺(tái)登記資料進(jìn)行關(guān)聯(lián)�。56 號(hào) 公告根據(jù)輔料包材風(fēng)險(xiǎn)進(jìn)行分級(jí)管理,并按需簡(jiǎn)化資料要求�。
5. 2019 年 12 月1日,《藥品管理法》生效�,藥品管理法第二十五條規(guī)定 “國(guó)務(wù)院藥品 監(jiān)督管理部門在審批藥品時(shí)����,對(duì)化學(xué)原料藥一并審評(píng)審批��,對(duì)相關(guān)輔料����、直接接觸藥 品的包裝材料和容器一并審評(píng),對(duì)藥品的質(zhì)量標(biāo)準(zhǔn)�����、生產(chǎn)工藝���、標(biāo)簽和說(shuō)明書一并核 準(zhǔn)” ���,在法律層面確立了原輔包與藥品關(guān)聯(lián)審評(píng)審評(píng)管理地位�。
6. 2020 年 7 月 1 日,《藥品注冊(cè)管理辦法》生效�����,第二章基本制度和要求第十四條再次 提出國(guó)家藥品監(jiān)督管理局建立化學(xué)原料藥�、輔料及直接接觸藥品的包裝材料和容器 關(guān)聯(lián)審評(píng)審批制度�����,在部門規(guī)章層面確立了原輔包與藥品關(guān)聯(lián)審評(píng)審評(píng)管理制度�。 其中第三章藥品上市注冊(cè)第三節(jié)關(guān)聯(lián)審評(píng)審評(píng)制度�,規(guī)定了原輔包與藥品制劑關(guān)聯(lián) 審評(píng)審批,由原輔包生產(chǎn)行業(yè)在原輔包登記平臺(tái)登記�����,藥品制劑注冊(cè)申請(qǐng)人提交注 冊(cè)申請(qǐng)時(shí)與平臺(tái)登記資料進(jìn)行關(guān)聯(lián);在藥品制劑注冊(cè)申請(qǐng)時(shí)�,選用未登記的原輔包的, 相關(guān)研究資料應(yīng)當(dāng)隨藥品制劑注冊(cè)申請(qǐng)一并申報(bào)���。藥品制劑注冊(cè)申請(qǐng)與已登記原輔 包進(jìn)行關(guān)聯(lián)��,藥品制劑獲得批準(zhǔn)時(shí)���,即表明其關(guān)聯(lián)的原輔包通過(guò)了技術(shù)審評(píng),登記 平臺(tái)標(biāo)識(shí)為“A” ;未通過(guò)技術(shù)審評(píng)或尚未與制劑注冊(cè)進(jìn)行關(guān)聯(lián)的標(biāo)識(shí)為“I” ����。藥審 中心向社會(huì)公示登記號(hào)、產(chǎn)品名稱、行業(yè)名稱��、生產(chǎn)地址等基本信息�����,供藥品制劑注 冊(cè)申請(qǐng)人選擇�����。
7. 2021 年4月����,為貫徹落實(shí)中共中央辦公廳、國(guó)務(wù)院辦公廳《關(guān)于深化審評(píng)審批制度改 革鼓勵(lì)藥品醫(yī)療器械創(chuàng)新的意見(jiàn)》(廳字〔2017〕42 號(hào))中關(guān)于藥品與藥用原輔料和 包裝材料關(guān)聯(lián)審批的要求����,同時(shí)根據(jù)新版《藥品管理法》、《藥品注冊(cè)管理辦法》���、
《總局關(guān)于調(diào)整原料藥�、藥用輔料和藥包材審評(píng)審批事項(xiàng)的公告》(2017 年第 146 號(hào)�, 以下簡(jiǎn)稱“146 號(hào)公告”)和《國(guó)家藥監(jiān)局關(guān)于進(jìn)一步完善藥品關(guān)聯(lián)審評(píng)審批和監(jiān)管 工作有關(guān)事宜的公告》(2019 年第 56 號(hào)�����,以下簡(jiǎn)稱“56 號(hào)公告”)關(guān)于原料藥、藥 用輔料和藥包材(以下簡(jiǎn)稱“原輔包”)在審批藥品制劑注冊(cè)申請(qǐng)時(shí)一并審評(píng)審批的 要求�,藥審中心起草了化學(xué)原料藥、藥用輔料及藥包材與藥品制劑共同審評(píng)審批管理 規(guī)定(征求意見(jiàn)稿)�,目前尚未定稿。
除以上法規(guī)和指導(dǎo)文件之外�����,2020-2021 年陸續(xù)發(fā)布了很多公告�、辦法及指南,對(duì)化學(xué)原 料藥登記要求����,繳費(fèi),已上市藥品中原輔包變更作出了相應(yīng)規(guī)定�。
建議:
建議擴(kuò)展登記適用范圍,對(duì)于原料藥中間體�、制劑中間體及生物技術(shù)原料藥也納入登記范圍。
建議原料藥的批準(zhǔn)日期與平臺(tái)轉(zhuǎn)“A”的日期開始計(jì)算���,此日期應(yīng)與《化學(xué)原料藥批準(zhǔn)通知書》載明的日期相同�����。同時(shí)��,建議盡快實(shí)現(xiàn)化學(xué)原料藥批準(zhǔn)通知書及其附件的“平臺(tái)打印”功能�����,以便申請(qǐng)人 / 登記人進(jìn)行化學(xué)原料藥的周期維護(hù)���。
按照《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》 第四章的要求�,變更納 入登記管理的輔料 / 藥包材����,變更后的輔料 / 藥包材尚未登記或登記狀態(tài)為 I 的,按照 重大變更管理�����。明確此類變更僅遞交路徑按照重大變更管理�����,但是相應(yīng)的研究工作���, 應(yīng)依據(jù)制劑的風(fēng)險(xiǎn)評(píng)估結(jié)果及《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》 /《已上市生物制品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》的要求提交變更支持性資料���, 而非將技術(shù)要求按登記狀態(tài)升級(jí)并按照重大變更管理��。
當(dāng)制劑申請(qǐng)同時(shí)關(guān)聯(lián)已登記的原輔包進(jìn)行關(guān)聯(lián)審評(píng)時(shí),建議原輔包 DMF 和制劑申請(qǐng)之 間是平行審評(píng)���,對(duì)原輔包的發(fā)補(bǔ)應(yīng)先于對(duì)制劑的發(fā)補(bǔ)或同時(shí)進(jìn)行發(fā)補(bǔ)��,這也是國(guó)際關(guān) 聯(lián)審評(píng)審批程序的慣例��,此舉可使藥品上市許可持有人同時(shí)準(zhǔn)備對(duì)于藥品各方面的提問(wèn)����,同時(shí)也可保證所有的疑慮都能及時(shí)解決��。
為保證患者用藥及藥品的持續(xù)供應(yīng)�����,建議根據(jù)《藥品注冊(cè)管理辦法》規(guī)定��,原輔 包的關(guān)聯(lián)審評(píng)同制劑審批類變更的審評(píng)時(shí)限一致����,即六十個(gè)工作日���,補(bǔ)充申請(qǐng)合并申報(bào)事項(xiàng)的,其審評(píng)時(shí)限為八十個(gè)工作日�����。
建議擴(kuò)展豁免輔料的范圍��,有些結(jié)構(gòu)簡(jiǎn)單的無(wú)機(jī)鹽或酸��,合成工藝簡(jiǎn)單�,作為 pH 調(diào) 節(jié)劑是可以豁免的,很多供應(yīng)商并不進(jìn)行平臺(tái)登記�����,但因其具備緩沖作用��,有時(shí)也用 作緩沖液 , 建議此種情形可按免登記進(jìn)行管理�。
建議考慮重新定義原輔包的平臺(tái)登記規(guī)則:
(1)同一申請(qǐng)人 / 登記人的不同生產(chǎn)廠所生產(chǎn)的原料藥、藥用輔料和藥包材��,如果使 用基本相同的生產(chǎn)工藝和相同的主要質(zhì)量標(biāo)準(zhǔn)��,可以在同一個(gè)登記號(hào)下進(jìn)行登記�����。 登記時(shí),應(yīng)注明所有相關(guān)的生產(chǎn)廠���。
(2)同一原料藥���、藥用輔料和藥包材的不同質(zhì)量等級(jí)����,如粒徑,藥包材材質(zhì)厚度等�����, 可以共用同一個(gè)登記號(hào)����。質(zhì)量標(biāo)準(zhǔn)可列入不同質(zhì)量等級(jí)的可接受標(biāo)準(zhǔn)。
(3)相同原理生產(chǎn)工藝的同種活性成分��,也可以根據(jù)其不同種質(zhì)量等級(jí)和適用范圍����, 按不同登記號(hào)管理�,而非只能登記一種最優(yōu)工藝����。例如行業(yè)有可能由于不同多晶型, 鹽或水合物等的需求�����,而使用不同的生產(chǎn)工藝����,其工藝并無(wú)優(yōu)劣之分。
建議考慮更改行業(yè)授權(quán)書模板�,納入多個(gè)制劑產(chǎn)品,同時(shí)�,不要求列明給藥途徑。
參考文獻(xiàn)
1.國(guó)內(nèi)外藥品技術(shù)指導(dǎo)原則體系對(duì)比研究 (藥學(xué)部分》�,國(guó)家藥品監(jiān)督管理局藥品審評(píng)中心,中國(guó)藥品監(jiān)督管理研究會(huì)����,藥品監(jiān)管研究國(guó)際交流專業(yè)委員會(huì),中國(guó)外商投資企業(yè)協(xié)會(huì)藥品研制和開發(fā)工作委員會(huì)(RDPAC)���,2022年11月

