在液體制劑研發(fā)中��,不可避免的問題是濾芯的選擇,凍干粉針通過濾芯過濾除菌��,大輸液和小水針通過濾芯過濾減菌��,口服溶液通過濾芯過濾控制可見異物等��。如果選擇合適的濾芯��?
依據(jù)最新版藥品GMP相關(guān)法律和法規(guī)��、歐盟GMP指南��、美國FDA《無菌工藝指南》及《除菌過濾技術(shù)及應(yīng)用指南》等相關(guān)法規(guī)和指南的要求��,選用的濾芯需要開展濾芯相容性研究��,濾芯與藥液相容性良好才可以在生產(chǎn)中使用��。濾芯相容性研究包括哪些呢��?
本文關(guān)于濾芯相容性研究��,進(jìn)行簡單的介紹��。濾芯相容性研究一般包括吸附試驗(yàn)��、細(xì)菌截留試驗(yàn)、化學(xué)兼容性試驗(yàn)��、可提取物或浸出物試驗(yàn)��、安全性評估等內(nèi)容��。
研發(fā)工作者需要考慮濾芯相容性的情況��,結(jié)合研發(fā)品種��,選擇合適的濾芯��。
本文結(jié)合濾芯相容性研究的5大板塊��,給出濾芯選擇的策略��。
一��、吸附試驗(yàn)
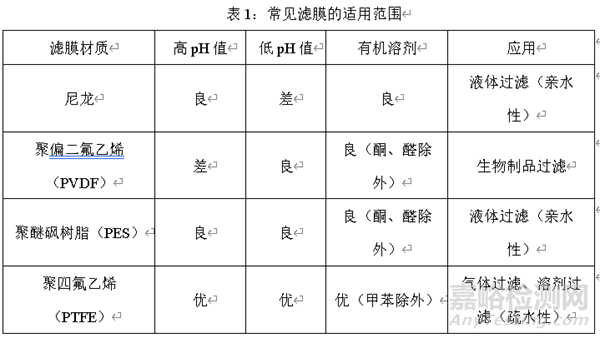
1��、在濾芯選擇時(shí)��,首先要充分考慮濾膜的適用范圍��,包括過濾溶液的pH值范圍��,過濾溶液的組成成分��,過濾器應(yīng)用的場景等��。
首先考慮藥液的pH值范圍��。
例如��,過濾的藥液pH值偏高��,不適宜選擇聚偏二氟乙烯的濾膜��,推薦使用聚四氟乙烯濾膜��。而過濾的藥液pH值偏低��,不宜選擇尼龍濾膜��,推薦使用聚四氟乙烯濾膜��。也就是說��,不論是在酸性環(huán)境還是堿性環(huán)境均適宜聚四氟乙烯濾膜��。
其次考慮藥液的組成��。過濾溶液組成成分不同,選擇的濾膜也不同��。含過濾溶液組成中有機(jī)相��,推薦使用聚四氟乙烯濾膜��。
最后��,在應(yīng)用場景方面��,尼龍和聚醚砜樹脂的過濾器適用于液體過濾��,聚四氟乙烯為疏水性濾膜適用于氣體過濾��,因?yàn)楦臑V膜具有疏水性��,進(jìn)行溶劑過濾時(shí)��,需要充分潤濕��。
下表將常見的濾膜適用范圍進(jìn)行梳理��,研發(fā)工作者可以結(jié)合開發(fā)的品種性質(zhì)對照下表進(jìn)行選擇:
2��、濾芯選擇要考慮濾芯材質(zhì)與原料��、輔料的相互作用��,充分考慮過濾器與藥液的兼容性��。
案例一:濾芯與原料相互作用
一般過濾器在過濾藥液過程中��,或多或少會(huì)吸附原料��。在廢棄一定藥液后��,過濾前后對主藥含量進(jìn)行檢測��,考察藥液廢棄不同體積后的含量變化��,以確定濾芯對原料的吸附影響��。
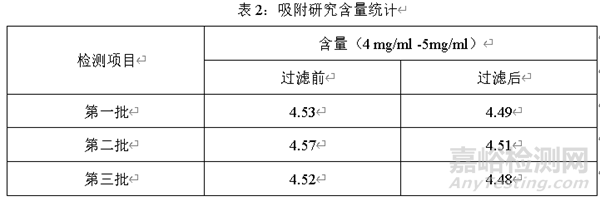
選擇三個(gè)批次藥液��,分別在過濾前和過濾后測定含量��,對比過濾前和過濾后含量的變化��,結(jié)果見下表
經(jīng)過三批過濾藥液前和過濾藥液后的含量可知��,過濾后的藥液含量與過濾前藥液的含量相比略微降低��,但是過濾前后含量無顯著變化,所以��,該過濾器對過濾藥液無吸附��。
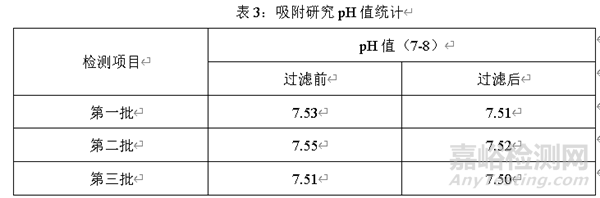
除了考慮藥液過濾前后含量的變化��,還應(yīng)考慮過濾前后藥液pH值變化��。選擇三個(gè)批次藥液��,分別在過濾前和過濾后測定pH值��,對比過濾前和過濾后pH值的變化��,結(jié)果如下
經(jīng)過三批過濾藥液前和過濾藥液后的pH值可知��,過濾后的藥液pH值與過濾前藥液的pH值相比略微降低��,但是過濾前后pH值無顯著變化��,通過pH值變化情況��,進(jìn)一步證實(shí)該過濾器對過濾藥液無吸附��。
生產(chǎn)過程中出現(xiàn)含量偏低的幾個(gè)考慮
如果過濾的藥液API含量低��,濾芯吸附的影響會(huì)相當(dāng)顯著。例如激素類藥物��,API在處方組成中占比小��,濾芯的吸附對API含量的影響極大��,在藥液正式灌裝前��,要充分考察藥液廢棄體積��。另外��,濾芯對API的吸附與濾芯的尺寸成正相關(guān)��,濾芯的尺寸越大��,濾芯吸附API達(dá)到飽和的量越大��;濾芯的尺寸越小��,濾芯吸附API達(dá)到飽和的量越小��。針對API在處方組成中占比小的情況��,可以選擇5英寸或者定制更小尺寸的濾芯��。
另外��,在生產(chǎn)過程中��,要充分考慮濾器在線滅菌的影響��,濾芯通過在線滅菌后��,會(huì)有部分水殘留在濾器中��,即使通過壓縮空氣或者氮?dú)獯祾?�,仍然不能把濾器的殘留水全部吹出��。這部分殘留水需要用藥液進(jìn)行充分的置換才能排出��。
生產(chǎn)管路在進(jìn)行清潔和蒸汽滅菌后��,管路降溫��,部分蒸汽會(huì)冷凝附著在管路內(nèi)壁形成殘留余水��,如果車間管路設(shè)計(jì)不合理,部分殘留余水無法通過排水口排出��,需要通過壓縮空氣或者氮?dú)膺M(jìn)行吹掃��,吹掃的時(shí)間和壓力不同��,殘留余水的量也不同��。需要控制管路吹掃時(shí)間和壓力��,確保濾芯和管路的殘留余水在可接受范圍內(nèi)��。
案例二:濾芯與輔料相互作用
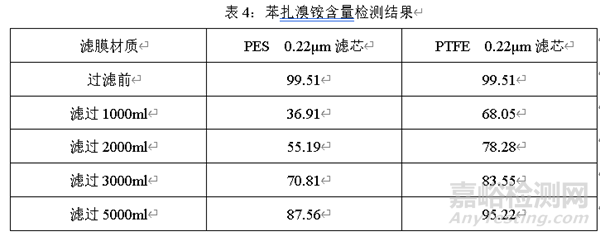
在研發(fā)過程中��,要考慮處方的某些成分與濾器的相容性��。過濾器在過濾藥液過程中��,可能會(huì)吸附某些輔料��,例如苯扎氯銨��、苯扎溴銨��。在過濾前檢測苯扎溴銨含量��,過濾不同體積��,檢測過濾不同體積后苯扎溴銨的含量��。
經(jīng)過上述考察可知��,不同材質(zhì)濾芯對苯扎溴銨均有吸附��。在廢棄相同體積后��,PTFE過濾的藥液苯扎溴銨含量比PES更高��,說明PTFE對苯扎溴銨的吸附與PES相比更小��。
在處方開發(fā)過程中��,要充分考慮特殊輔料(如:苯扎溴銨��、苯扎氯銨等)與濾芯的相容性��。
除了吸附的影響��,過濾器不得因與產(chǎn)品發(fā)生反應(yīng)��、釋放物質(zhì)而對產(chǎn)品質(zhì)量產(chǎn)生不利影響��。
總之,在濾芯吸附試驗(yàn)板塊��,濾芯的選擇��,首先要考慮濾芯的適用范圍��,其次是濾芯與原料��、輔料以及藥液的相容性��。
二��、影響細(xì)菌截留的因素
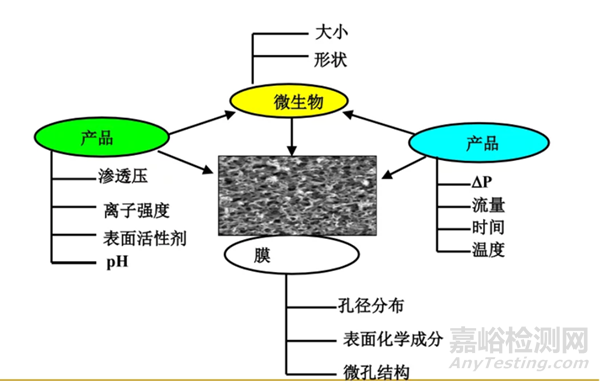
首先是微生物的性狀和大?�?�;
其次是藥液的性質(zhì)��,包括滲透壓��、離子強(qiáng)度��、pH值��、表面活性劑占比��、有機(jī)成分占比��、抑菌或殺菌成分的占比等��;
然后是濾膜的性質(zhì)��,包括孔徑分布��、表面化學(xué)成分��。微孔結(jié)構(gòu)等��;
最后是藥液通過濾膜的壓差��、時(shí)間��、溫度��、批量等��。
影響細(xì)菌截留的因素詳見下圖:
圖5:影響細(xì)菌截留的因素
三��、細(xì)菌截留的研究方式
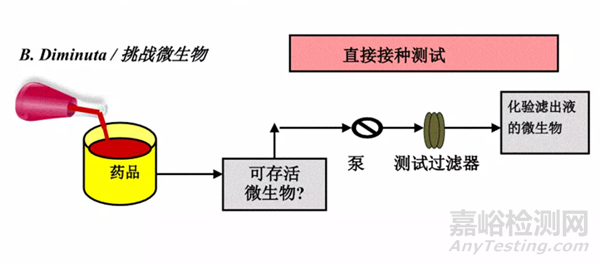
細(xì)菌截留的研究方式一般有兩種:
直接法和間接法��。
在開展細(xì)菌截留研究前��,要先確認(rèn)藥液對細(xì)菌的影響。確認(rèn)藥液對細(xì)菌的影響分為4步:
第一步
在生產(chǎn)工藝條件下,將挑戰(zhàn)菌直接加入到產(chǎn)品中,并在工藝條件下進(jìn)行培養(yǎng)��。
第二步
檢測產(chǎn)品中挑戰(zhàn)菌的存活率��。
第三步
根據(jù)挑戰(zhàn)菌在測試液和對照液中的LRV值不同,可以將測試液分成殺菌性產(chǎn)品��、非殺菌性產(chǎn)品和中度殺菌性產(chǎn)品三種��。
第四步
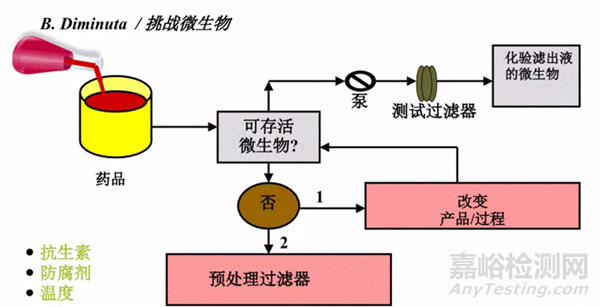
如果產(chǎn)品為非殺菌性產(chǎn)品,則細(xì)菌挑戰(zhàn)測試的方式為直接將挑戰(zhàn)菌加入產(chǎn)品中進(jìn)行挑戰(zhàn);
如產(chǎn)品為中度殺菌性產(chǎn)品或殺菌性產(chǎn)品,則需要建立濾芯或?yàn)V膜的沖洗方案,驗(yàn)證產(chǎn)品殘留對細(xì)菌生存性的影響,并選擇合適的替代液代替客戶產(chǎn)品進(jìn)行細(xì)菌挑戰(zhàn)測試��。
圖2:細(xì)菌截留研究方式——直接法
圖3:細(xì)菌截留研究方式——間接法
判斷標(biāo)準(zhǔn)
1��、在工藝時(shí)間內(nèi)活菌數(shù)下降<1log,確定測試藥液為非殺菌性產(chǎn)品��。
2��、1h內(nèi)活菌數(shù)下降<1log��,但是在工藝時(shí)間內(nèi)活菌數(shù)下降≥1log,確定測試藥液為中度殺菌性產(chǎn)品��。
3��、1h內(nèi)活菌數(shù)下降>1log,確定測試藥液為殺菌性產(chǎn)品��。
4��、恢復(fù)生長的菌落數(shù)必須在 20~200 cfu 才可作為有效計(jì)數(shù)結(jié)果��。
四��、細(xì)菌截留試驗(yàn)需要考慮的方面
1��、在除菌過濾驗(yàn)證中使用濾膜還是濾器,取決于驗(yàn)證的目的��。如果微生物截留試驗(yàn)的目的是驗(yàn)證過濾工藝中特定膜材的細(xì)菌截留效能,那么使用濾膜是能滿足需要的��。微生物截留試驗(yàn)中所用的濾膜必須和實(shí)際生產(chǎn)中所用過濾器材質(zhì)完全相同,并應(yīng)包括多個(gè)批次(通常三個(gè)批次)��。其中至少應(yīng)有一個(gè)批次為低起泡點(diǎn)(低規(guī)格)濾膜��。
2��、為了在微生物挑戰(zhàn)試驗(yàn)中實(shí)施最差條件,一般需要使用完整性測試的數(shù)值非常接近過濾器生產(chǎn)商提供的濾器完整性限值的濾膜(例如不高于標(biāo)準(zhǔn)完整性限值的110%) ��。如果在驗(yàn)證中沒有使用低起泡點(diǎn)濾膜,那么在實(shí)際生產(chǎn)中所使用的標(biāo)準(zhǔn)溶液濾膜/芯起泡點(diǎn)值,必須高于驗(yàn)證試驗(yàn)中實(shí)際使用的濾膜的最小起泡點(diǎn)值��。
3��、成分相容��、只有濃度不同的系列產(chǎn)品可以通過挑戰(zhàn)極限濃度和接受中間一組濃度來驗(yàn)證��。
4、一般來說重復(fù)使用過濾器是不實(shí)際的��,如果重復(fù)使用除菌級過濾器��,應(yīng)該給出充足里有并且重復(fù)使用參數(shù)經(jīng)過充分論證��。
結(jié)合細(xì)菌截留試驗(yàn)的研究內(nèi)容��,關(guān)于濾膜孔徑選擇和濾芯組合系統(tǒng)選擇��,本文推薦如下:
關(guān)于濾膜孔徑的選擇��,在這里��,要先科普一下��,世界上最小的細(xì)菌是缺陷型假單胞菌(直徑大約為0.3—0.4微米��,長度0.6—1.0微米)��。在除菌過濾驗(yàn)證中��,缺陷型假單胞菌必須是單一的��、分散的細(xì)胞��,才能作為細(xì)菌截留試驗(yàn)的標(biāo)準(zhǔn)挑戰(zhàn)微生物��。所以只有過濾器孔徑小于缺陷型假單胞菌的尺寸時(shí)��,才能保證對最小微生物的截留能力��。一般情況��,除菌過濾工藝應(yīng)選用0.22微米(更小孔徑或相同過濾效力)的除菌級過濾器��。
濾芯組合一般分為兩種:序列過濾系統(tǒng)和冗余過濾系統(tǒng)��。
通常通過兩個(gè)或以上相同或遞減孔徑的過濾方式��,統(tǒng)稱為序列過濾系統(tǒng)��。例如:0.45μm過濾器+0.22μm過濾器��。序列過濾系統(tǒng)中��,如果在最終除菌過濾器前增加一個(gè)除菌級過濾器��,并且確保兩個(gè)過濾器之間無菌��,以及控制過濾前介質(zhì)的微生物污染水平一般小于等于10cfu/100ml��,這種情況下稱為冗余過濾系統(tǒng)。例如:0.45μm過濾器+0.22μm過濾器+0.22μm過濾器��。
總之��,通過細(xì)菌截留試驗(yàn)研究和論證��,除菌過濾工藝應(yīng)選用0.22微米(更小孔徑或相同過濾效力)的除菌級過濾器��,除菌過濾工藝一般選擇冗余過濾系統(tǒng)��,達(dá)到更高的無菌保障水平��。
五��、化學(xué)兼容性試驗(yàn)
根據(jù)國內(nèi)外GMP相關(guān)條款要求:與藥品直接接觸的濾器不得與藥液發(fā)生化學(xué)反應(yīng)��、吸附藥品或者向藥液中釋放物質(zhì)��。
化學(xué)兼容性試驗(yàn)要包括濾芯的全部組成部件(包括濾膜��,濾殼外骨架��、端蓋支撐部件��,密封圈等)��。
在最差實(shí)際過濾工藝條件下��,考察對濾芯過濾性能的影響��,包括2個(gè)方面:
1是藥液是否影響濾芯過濾性能��,
2是濾芯是否影響藥液性質(zhì)��。
過濾工藝要考慮待過濾介質(zhì)性質(zhì)��、過濾溫度��、接觸時(shí)間��、過濾批量��、過濾批次等��?�?紤]過濾介質(zhì)性質(zhì)��,例如過濾介質(zhì)是否含有乙醇等有機(jī)相��,有機(jī)相的組成比例,試驗(yàn)過程中的過濾溫度應(yīng)達(dá)到或者超過生產(chǎn)過程的最高溫度��,過濾時(shí)間應(yīng)達(dá)到或者超過實(shí)際生產(chǎn)過程的最長工藝時(shí)間��。
化學(xué)兼容性試驗(yàn)檢測項(xiàng)目一般包括:
1.濾芯
過濾器接觸待過濾介質(zhì)前后的目視檢查��,要求外觀無損壞��、變形和軟化現(xiàn)象��,起泡點(diǎn)值大于或者等于3200mbar(測試液體為水)��,擴(kuò)散流值小于等于10ml/min/5in@0.28Mpa(測試液體為水)��,浸泡前后相對偏差不超過10%��。
2.支撐部門
過濾器接觸待過濾介質(zhì)前后目視檢查��,要求外觀無破損和軟化現(xiàn)象��。
3.濾膜
接觸過濾介質(zhì)前和接觸過濾介質(zhì)后��,要求外觀無破損和軟化現(xiàn)象��,過濾過程中流速變化��,一般要求不超過20%;濾膜重量是否變化��,重量變化一般不超過5%��;厚度是否變化��,厚度變化一般不超過5%��;濾膜拉伸強(qiáng)度的變化��,拉伸變形率一般不超過5%��;濾膜電鏡掃描確認(rèn)無明顯差異��。
4.密封圈
接觸過濾介質(zhì)前和接觸過濾介質(zhì)后��,要求外觀無變形��、溶脹和軟化現(xiàn)象��,密封圈內(nèi)徑變化要求浸泡前后相對偏差在±2%之內(nèi)��,重量要求浸泡前后相對偏差在±2%之內(nèi)��。
5.藥液
接觸過濾介質(zhì)前和接觸過濾介質(zhì)后��,要求無肉眼可見的濾芯脫落物��。
特別需要注意的是��,濾器接觸藥液后��,藥液中可能存在部分顆粒堵塞濾膜孔��,進(jìn)而導(dǎo)致浸泡后濾芯的泡點(diǎn)和過濾阻力增加��,所以��,過濾后起泡點(diǎn)相對偏差不設(shè)置上限��。
在化學(xué)兼容性部分��,要考慮濾芯的過濾阻力大小��、濾膜厚度��、拉伸強(qiáng)度��,結(jié)合車間生產(chǎn)線的實(shí)際情況��,選擇筒式或者囊式濾芯��。
六、可提取物或浸出物試驗(yàn)
根據(jù)NMPA的GMP(2010版)提出了如下要求:生產(chǎn)設(shè)備不得對藥品質(zhì)量產(chǎn)生任何不利影響��。因此要關(guān)注生產(chǎn)設(shè)備(例如過濾器)與工藝溶液接觸會(huì)后��,其結(jié)構(gòu)可能發(fā)生改變��,進(jìn)而導(dǎo)致過濾器組分遷移至藥品中��。
可提取物是在極端條件下(例如有機(jī)溶劑��、極端高溫��、pH值��、接觸時(shí)間)��,可以從過濾器提取出的化學(xué)物質(zhì)��。浸出物是在正常工藝條件和試機(jī)藥品中��,從過濾器遷移出來的化合物��。
使用最長過濾時(shí)間��、最高過濾溫度��、最多次蒸汽滅菌循環(huán)��、增加伽瑪輻射的次數(shù)和劑量都可能會(huì)增加可提取物水平��。
可提取物反映了浸出物的最大可能��。
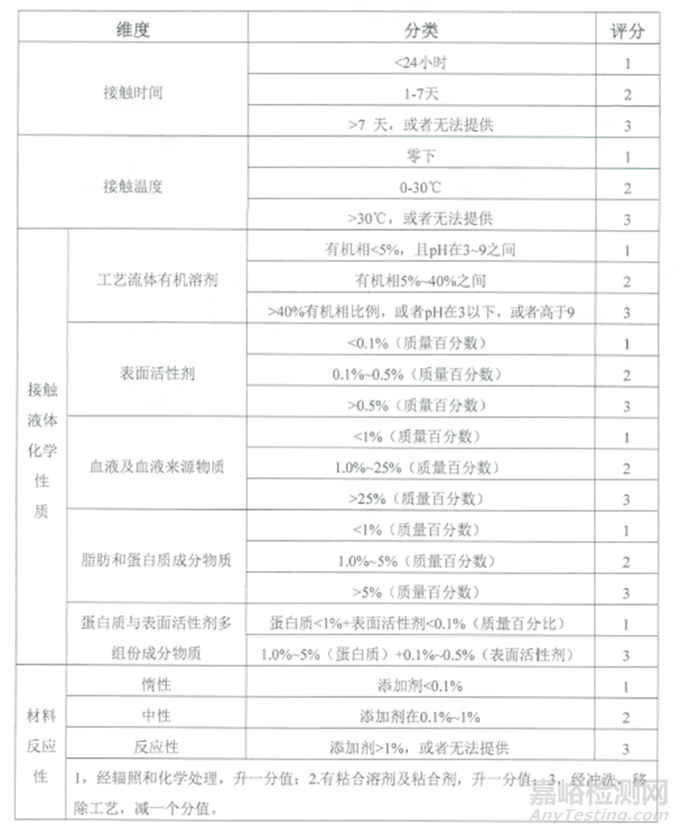
可提取物試驗(yàn)可以用靜態(tài)浸泡或循環(huán)流動(dòng)的方法��,其影響因素包括接觸時(shí)長��、接觸溫度��、溶液類型和組件材料活性��。
表1:可提取物維度��、分類及評分匯總表
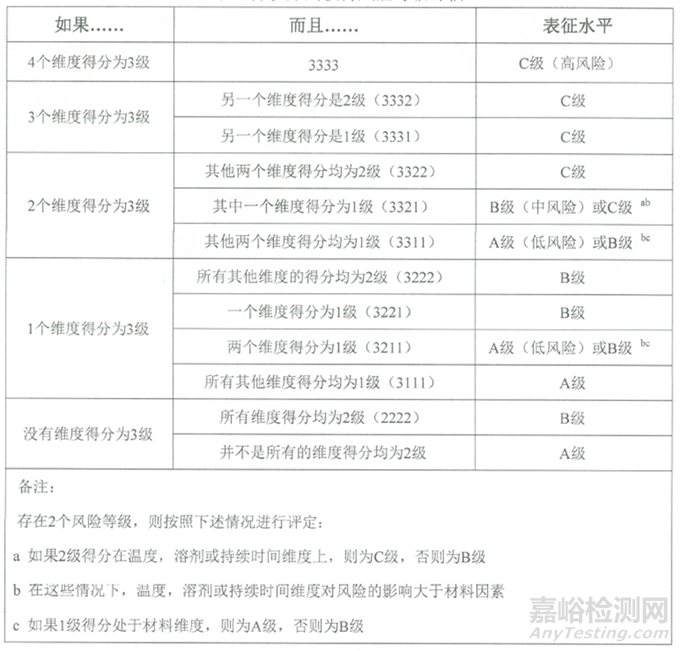
根據(jù)接觸時(shí)長��、接觸溫度��、溶液類型和組件材料活性等維度的評分��,評估出提取物研究的風(fēng)險(xiǎn)等級��。提取物研究的風(fēng)險(xiǎn)等級詳見下表:
表2:提取物研究的風(fēng)險(xiǎn)等級評估表
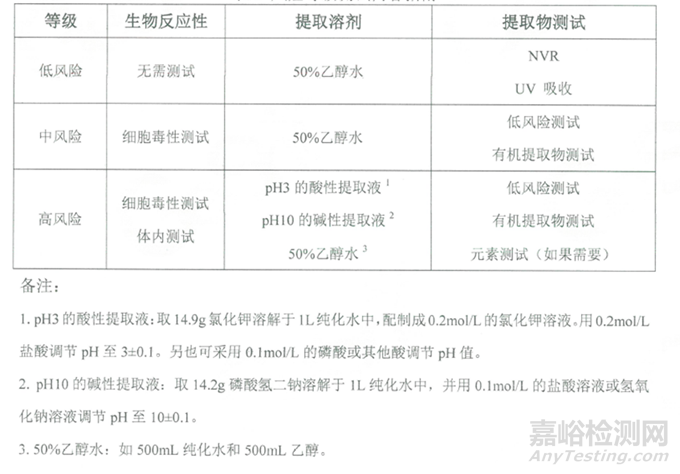
根據(jù)提取物評估的風(fēng)險(xiǎn)等級��,開展相應(yīng)的研究,不同等級對應(yīng)的提取物測試匯總表如下:
表3:不同等級對應(yīng)的提取物測試匯總表
提取物或浸出物研究包括:
1��、 不揮發(fā)物(NVR)檢測可提取物中非揮發(fā)物
2��、 紫外可見吸收光譜(UV-VIS)檢測發(fā)色物質(zhì)的官能團(tuán)
3��、 傅里葉變換紅外光譜法(FTIR)檢測聚合物或寡聚物的官能團(tuán)
4��、 反相高效液相色譜(RP-HPLC)檢測溶劑��、單體和添加劑
5��、 氣相色譜-質(zhì)譜(GC-MS)揮發(fā)及半揮發(fā)物
用于試驗(yàn)的過濾器盡量不進(jìn)行預(yù)沖洗��。在可提取物和浸出物試驗(yàn)部分��,要結(jié)合處方組成(有機(jī)相比例)��,工藝參數(shù)(過濾溫度��、過濾時(shí)間��、過濾體積和批量)��,滅菌方式(在線滅菌還是離線滅菌)等因素��,選擇適宜的濾芯��。
七��、安全性評估
在完成可提取物或者浸出物試驗(yàn)后��,應(yīng)針對過濾器可提取物或浸出物的種類和含量��,結(jié)合藥品最終劑型中的濃度��、劑量大小��、給藥時(shí)間��、給藥途徑等對結(jié)果進(jìn)行安全性評估��,以評估可提取物和浸出物是否存在安全性風(fēng)險(xiǎn)��。
參考文獻(xiàn)
1.國家食品藥品監(jiān)督管理局.藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂).2011.
2.US Food and Drug Administration, Current Good Manufacturing Practice for Finished Pharmaceuticals, Equipment Construction, 21 CFR Part 211.65(a), 2019.
3.European Commission, EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Premise and Equipment, Chapter 3.39, 2015.
4.U.S. Department of Health and Human Services, Food and Drug Administration, Guidance for Industry, Container Closure Systems for Packaging Human Drugs and Biologics, May 1999.
5.Parenteral Drug Association (PDA), Sterilizing filtration of liquids technical report No.26, 2008
6.國家食品藥品監(jiān)督管理局藥品認(rèn)證管理中心.無菌藥品:藥品GMP指南. 2011.
7.國家藥品監(jiān)督管理局.除菌過濾技術(shù)及應(yīng)用指南(2018年第85號通告) .2018-07-31.
8.國家藥品監(jiān)督管理局藥品審評中心.化學(xué)藥品注射劑生產(chǎn)所用的塑料組件系統(tǒng)相容性研究技術(shù)指南(試行)(2020年第33號通告).2020-10-21
9.Ding, W., et al., "Standardized Extractables Testing Protocol for Single-Use Systems in Biomanufacturing," Pharmaceutical Engineering, Vol. 34, No.4, 2014.
10 United States Pharmacopeial Convention, Plastic Components and Systems Used to Manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products, <665>,2022.
1.American Society for Testing and Material. Standard Test Method for Determining Bacterial of Membrane Filters Utilized for Liquid Filtration. 1993 ASTM Standard and Environmental Microbiology,Second Edition, Designation F838-83, pp 50-55.
2.Center for Drugs and Biologics and Office of Regulatory Affairs, Food and Drug Administration. Guideline on Sterile Drug Products Produced by Aseptic Processing. September2004.
3.PDA Journal of Pharmaceutical Science and Technology: Technical Report No. 26, Sterilizing Filtration of Liquids, March 1998.
4.國家食品藥品監(jiān)督管理局,藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂) [S].2011
5.國家食品藥品監(jiān)督管理局藥品認(rèn)證管理中心,無菌藥品:藥品GMP指南[S].2011
6.國家藥品監(jiān)督管理局.除菌過濾技術(shù)及應(yīng)用指南(2018年第85號通告) [2].2018-07-31
7.國家藥品監(jiān)督管理局,除菌過濾技術(shù)及應(yīng)用指南��,2018
8.Parenteral Drug Association(PDA), Sterilizing filtration of liquids technical report No.26,2008
9.General Information Chapter<1227>,Validation of Microbial Recovery from PhamacopeialArticles, USP 28,USPC,Inc.,Rockville,MD,2005,p. 2752
10.American Society for Testing and Materials (ASTM) Standard Test Method for Determining Bacterial Retention of Membrane Filters Utilized for Liquid Filtration F838-05,pp1-6,ASTM,WestConshohocken,PA,2005.
1.國家食品藥品監(jiān)督管理局.藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂).2011.
2.US Food and Drug Administration, Current Good Manufacturing Practice for Finished Pharmaceuticals, Equipment Construction, 21 CFR Part 211.65(a), 2019.
3.European Commission, EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Premise and Equipment, Chapter 3.39, 2015.
4.U.S. Department of Health and Human Services, Food and Drug Administration, Guidance for Industry, Container Closure Systems for Packaging Human Drugs and Biologics, May 1999.
5.Parenteral Drug Association (PDA), Sterilizing filtration of liquids technical report No.26, 2008
6.國家食品藥品監(jiān)督管理局藥品認(rèn)證管理中心.無菌藥品:藥品GMP指南. 2011.
7.國家藥品監(jiān)督管理局.除菌過濾技術(shù)及應(yīng)用指南(2018年第85號通告) .2018-07-31.
8.國家藥品監(jiān)督管理局藥品審評中心.化學(xué)藥品注射劑生產(chǎn)所用的塑料組件系統(tǒng)相容性研究技術(shù)指南(試行)(2020年第33號通告).2020-10-21
9.Ding, W., et al., "Standardized Extractables Testing Protocol for Single-Use Systems in Biomanufacturing," Pharmaceutical Engineering, Vol. 34, No.4, 2014.
10 United States Pharmacopeial Convention, Plastic Components and Systems Used to Manufacture Pharmaceutical Drug Products and Biopharmaceutical Drug Substances and Products, <665>,2022.
11. Container Closure Systems for Packaging Human Drugs and Biologics
U.S. Depaitment of Health and Human Services Food and Drug Administration Center for Drug
Evaluation and Research (CDER) & Center for Biologics Evaluation and Research (CBER). May
1999
12. 國家食品藥品監(jiān)督管理總局��,化學(xué)藥品與彈性體密封件相容性研究技術(shù)指導(dǎo)原則��,2016
13. GB/T 34244-2017 液體除菌用過濾芯技術(shù)要求