方法: 采用正交設(shè)計法,以脆碎度�、崩解時限�����、黏沖情況���、在酸中60min溶出度的綜合評分為指標(biāo)優(yōu)化處方�。
結(jié)果: 最佳壓片處方為微晶纖維素20%���、硬脂酸鎂2%���、交聯(lián)聚維酮7%�����、蘭索拉唑腸溶微丸15%�。3批樣品脆碎度�、崩解時限���、含量和含量均勻度均符合要求; 40℃放置6個月脆碎度�、崩解時限、含量�、有關(guān)物質(zhì)和溶出度均無明顯變化。
結(jié)論: 使用正交實驗優(yōu)化處方制備的蘭索拉唑腸溶口崩片質(zhì)量穩(wěn)定���、可控�����。
關(guān)鍵詞 蘭索拉唑; 腸溶口崩片; 微丸壓片; 正交實驗; 穩(wěn)定性
蘭索拉唑(lansoprazole)臨床主要用于治療胃及十二指腸潰瘍�����,其療效較好�,不良反應(yīng)少�����,抑酸作用是奧美拉唑的2倍[1]。目前�����,市場上國產(chǎn)的蘭索拉唑制劑主要有腸溶片和腸溶膠囊2種劑型�����,而微丸片只有日本武田公司生產(chǎn)的腸溶蘭索拉唑口崩片,商品名為普托平。腸溶制劑主要有單單元制劑和多單元制劑2種�����,大多數(shù)的腸溶片劑都是單單元制劑�,因此藥物的作用和吸收容易受到消化物遷移的影響���,而多單元制劑能彌補這個缺點�����。但普通的膠囊體積較大���,患者不易吞咽���,用藥的順應(yīng)性較低; 再者�,有些藥物使用時需要調(diào)整劑量�����,而片劑相較膠囊給藥更加靈活[2-3]。蘭索拉唑口崩片是口腔內(nèi)快速崩解的質(zhì)子泵抑制�,該藥放入口腔后�,會在30s內(nèi)崩解為無數(shù)包有腸溶衣的小丸微粒�,每個微粒由幾層結(jié)構(gòu)組成,這樣可以抵抗胃蠕動所造成的擠壓�����,從而保證藥效不受影響[4-10]。
本研究是在前期研究的基礎(chǔ)上�����,采用流化床上藥技術(shù)制備蘭索拉唑載藥丸芯�,流化床包衣技術(shù)包隔離衣和腸溶衣,并加其他輔料與腸溶微丸混合壓片���。以蘭索拉唑腸溶微丸�、稀釋劑微晶纖維素�、崩解劑交聯(lián)聚維酮、潤滑劑硬脂酸鎂���,采用正交設(shè)計法���,選擇片劑崩解時限、脆碎度�����、黏沖情況和在酸中60min溶出度的綜合評分作為評價指標(biāo)優(yōu)化處方�,最終制備成蘭索拉唑腸溶口崩片,同時考察制劑質(zhì)量及穩(wěn)定性�����。
材料與方法
1 藥品與試劑
蘭索拉唑(桂林華信制藥有限公司���,批號: 56722035155); 蔗糖丸芯0.250~0.355mm(杭州高成生物營養(yǎng)技術(shù)有限公司���,批號: 20231001); 堿式碳酸鎂(Sudeep Pharma Pvt. Ltd. 公司,批號: 21K/ML/010); 羥丙甲纖維素E5(安徽山河藥用輔料股份有限公司�,批號: 210126); 尤特奇L30D-55(德國贏創(chuàng)公司,批號: B210403204); 尤特奇NE30D(德國贏創(chuàng)公司�,批號: B220501540); 單硬脂酸甘油酯(GAT TEFOSSE SAS公司,批號: 195281); 檸檬酸三乙酯(江西阿爾法高科藥業(yè)有限公司���,批號: 20230401); 紅氧化鐵(寧波一品生物技術(shù)有限公司�,批號: 230215); 聚山梨酯80(湖北葛店人福藥用輔料有限責(zé)任公司���,批號: F104C220101); 微晶纖維素(micro crystalline cellulose���,MCC) 102(湖州展望藥業(yè)有限公司���,批號: 20221001); 甘露醇(羅蓋特公司,批號: E257H); 交聯(lián)聚維酮XL-10(ISP Chemicals LL公司���,批號: 060224033); 阿斯帕坦(湖南九典宏陽制藥有限公司�,批號: TF67230201); 無水檸檬酸(Merck KGaA公司�,批號: K52745002); 草莓香精[曼氏(上海)香精香料有限公司,批號: E21088687]; 硬脂酸鎂(安徽山河藥用輔料股份有限公司���,批號: 200354)���。
2 儀器
LBL1型流化床制粒包衣機(jī)(重慶市科旭制藥機(jī)械制造有限公司); BSL25型料斗混合機(jī)(重慶瀚威迪科技有限公司); ZP10A型旋轉(zhuǎn)式壓片機(jī)(北京新龍立科技有限公司); ESJ1004B型天平(沈陽神宇龍騰天平有限公司); YD35型片劑硬度儀(天津市天大天發(fā)科技有限公司); KB1型口崩片崩解儀(天津市天大天發(fā)科技有限公司); FOCS ZJ2型溶出儀(安捷倫科技有限公司); UV2600型紫外分光光度計(日本島津公司); S210型pH測定計(賽多利斯科學(xué)儀器有限公司); 1260ImfinityⅡ型高效液相色譜儀(美國安捷倫科技有限公司)。
3 腸溶片的制備方法
3.1 載藥丸芯
將羥丙甲纖維素E5(15%)分散在80℃純化水中�,加入堿式碳酸鎂(10%)和蘭索拉唑(29%),攪拌均勻���,得上藥混懸液�����。將蔗糖丸芯(46%)置于流化床中�����,待物料溫度達(dá)到38℃以上開始噴上藥混懸液���,噴完后保持同樣的參數(shù)干燥10min���。分別過30和55目篩,收集30~55目的微丸�����,得載藥丸芯。
3.2 隔離衣層將羥丙甲纖維素E5(8.3%)在80℃純化水中分散攪拌�����,冷卻至澄清; 加入滑石粉(8.3%)�����,攪拌成混懸液�。將載藥丸芯(83.4%)置于流化床中,待物料溫度達(dá)到38℃以上開始噴液���,噴完后保持同樣的參數(shù)干燥10min���。分別過30和55目篩,收集30~55目的微丸�����,得隔離衣微丸�����。
3.3 腸溶衣層將聚山梨酯80(1%)�����、檸檬酸三乙酯(2%)和單硬脂酸甘油酯(1%)加入純化水中攪拌均勻,再加入尤特奇L30D-55(12.7%)水分散體中���,攪拌均勻,得混懸液���。將隔離衣微丸(83.3%)置于流化床中�����,待物料溫度達(dá)到25℃以上開始噴混懸液�,噴完后保持同樣的參數(shù)干燥10min�����。干燥后分別過30和45目篩�����,收集30~50目的微丸���,得到蘭索拉唑腸溶微丸���。
3.4 腸溶微丸壓片及處方優(yōu)化將蘭索拉唑腸溶微丸���、MCC102、甘露醇�、交聯(lián)聚維酮XL-10、阿斯帕坦(0.5%)�����、檸檬酸(1%)���、草莓香精(0.5%)和硬脂酸鎂置料斗混合機(jī)中混合�����,再使用壓片機(jī)壓片�����,得腸溶口崩片�。
由于腸溶口崩片的壓片輔料成分性質(zhì)不同�、在處方中的作用不同,所以各成分的合理配比是制成優(yōu)良處方的關(guān)鍵���。前期研究中,對各輔料在處方中的作用進(jìn)行了分析,對腸溶口崩片制劑壓片工藝影響較大的輔料有MCC���、交聯(lián)聚維酮、硬脂酸鎂等。在前期研究的基礎(chǔ)上�����,選取MCC(A)、硬脂酸鎂(B)�����、交聯(lián)聚維酮(C)���、蘭索拉唑腸溶微丸(D)作為4個因素�����,確定正交實驗水平(見表1)�����。以稀釋劑甘露醇補足剩余處方量�����,并根據(jù)口崩片制劑應(yīng)用特點�,分別以崩解時限、脆碎度�、黏沖情況和在酸中60min的溶出度(見表2),按照L9(34)正交表進(jìn)行實驗�,對各指標(biāo)結(jié)果加權(quán)后進(jìn)行綜合評分[綜合評分=崩解時限評分×0.3+脆碎度評分×0.2+黏沖評分×0.2+在酸中60min溶出度×0.3]。
4 崩解時限測定方法
取本品���,按照《中華人民共和國藥典》2020年版通則0921進(jìn)行檢查�,應(yīng)在60s內(nèi)全部崩解并通過篩網(wǎng)�����,重復(fù)測定6片�。
5 脆碎度測定方法
取本品約0.65g,吹去片劑脫落的粉末�����,精密稱重���,置脆碎度儀圓筒中�����,轉(zhuǎn)動100次�����,取出�����,同法除去粉末���,精密稱重,減失重量不得超過1%�,且不得檢出斷裂、龜裂及粉碎的片�����。
6 體外釋放度測定方法取本品按照《中華人民共和國藥典》2020年版通則0931第二法(槳法)測定���,以0.1mol·L-1鹽酸溶液為溶出介質(zhì)Ⅰ�,在轉(zhuǎn)速為75r·min-1下進(jìn)行溶出實驗����,1h后取溶液25mL��,濾過�,棄去初濾液10mL�����,取續(xù)濾液作為供試品溶液Ⅰ��,立即將37℃磷酸鹽緩沖液425mL緩慢加入溶出杯中��,迅速用2mol·L-1鹽酸溶液或2mol·L-1氫氧化鈉溶液調(diào)節(jié)pH值至6.80±0.05�,作為溶出介質(zhì)Ⅱ,繼續(xù)溶出45min�,取溶液25mL,濾過�,棄去初濾液10mL,取續(xù)濾液加溶出介質(zhì)Ⅱ稀釋制成1mL中約含蘭索拉唑16μg的溶液��,作為供試品溶液Ⅱ����。另精密稱取蘭索拉唑?qū)φ掌?0mg,置50mL量瓶中,加甲醇溶解并稀釋至刻度�,作為對照品貯備液,精密量取對照品貯備液適量�����,加0.1mol·L-1鹽酸稀釋至1mL中含2.4μg的對照品溶液��,作為對照品溶液Ⅰ�。取供試品溶液Ⅰ和對照品溶液Ⅰ,照紫外分光光度法�,在332nm波長處分別測定吸光度,計算每片的釋放量; 再精密量取對照品貯備液1mL�����,置100mL量瓶中�����,加溶出介質(zhì)Ⅱ稀釋至刻度�����,搖勻���,作為對照品溶液Ⅱ�,取供試品溶液Ⅱ和對照品溶液Ⅱ�,照紫外分光光度法,在286nm波長處分別測定吸光度�,計算每片的釋放量。
7 含量均勻度測定方法
取本品1片����,置100mL量瓶中,加入0.1mol·L-1氫氧化鈉溶液30mL�����,超聲使片劑完全崩解并使蘭索拉唑溶解��,加乙腈65mL����,室溫放置1h,加乙腈稀釋至刻度�,搖勻,離心���,取上清液濾過��,棄去初濾液5mL����,精密量取續(xù)濾液適量,用0.1mol·L-1氫氧化鈉溶液-乙腈(3∶7)稀釋制成每1mL中含蘭索拉唑12μg的溶液���,作為供試品溶液���。另取蘭索拉唑?qū)φ掌愤m量,精密稱定��,同法操作制成1mL溶液中含蘭索拉唑?qū)φ掌?2μg的溶液�。照紫外分光光度法,在294nm波長處測定吸光度�����,計算含量�。文章內(nèi)容由凡默谷小編查閱文獻(xiàn)選取,排版與編輯為原創(chuàng)�����。如轉(zhuǎn)載�,請尊重勞動成果,注明【來源:凡默谷公眾號】���。
結(jié)果
1 壓片處方優(yōu)化結(jié)果
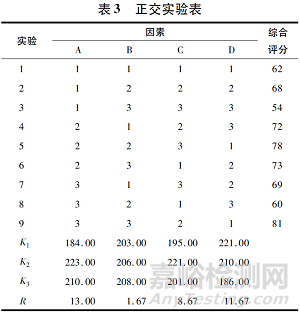
直觀分析結(jié)果見表3�。
各因素對指標(biāo)影響的大小次序為A>D>C>B�����,即MCC用量對崩解時限���、在酸中的溶出度�、脆碎度�、黏沖情況影響最顯著,R值為13.00; 蘭索拉唑腸溶微丸和交聯(lián)聚維酮用量對腸溶口崩片各性能的影響次之�����,R值分別為11.67和8.67; 而硬脂酸鎂對制劑的影響最小���,R值為1.67���。最佳組合為A2B3C2D1,即MCC為20%����、硬脂酸鎂為2%�、交聯(lián)聚維酮為7%�、蘭索拉唑腸溶微丸為15%。
2 質(zhì)量考察
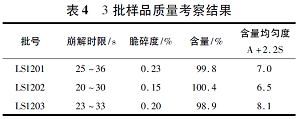
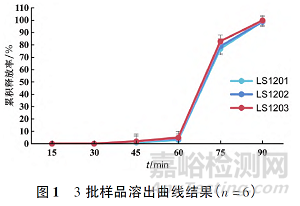
按照優(yōu)化處方連續(xù)制備3批樣品�����,分別測定崩解時限�、脆碎度、含量���、含量均勻度�����、溶出曲線��,結(jié)果見表4和圖1�。
3 穩(wěn)定性考察
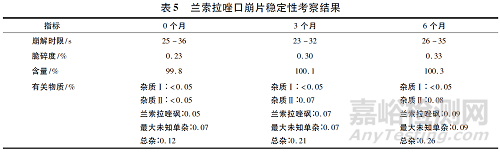
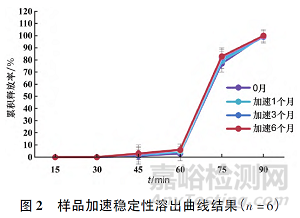
將制得的蘭索拉唑口崩片進(jìn)行鋁塑包裝����,置于40 ℃, 75%RH 條件下6 個月進(jìn)行穩(wěn)定性考察��,結(jié)果見表5 和圖2。結(jié)果表明����,40 ℃放置6 個月樣品脆碎度�����、崩解時限���、含量�、有關(guān)物質(zhì)和溶出度均無明顯變化����,產(chǎn)品穩(wěn)定性較好。
討論
本研究采用流化床上藥法制備載藥丸芯�����,流化床包衣得到腸溶微丸���,最后將腸溶微丸與其他輔料混合��,壓制成蘭索拉唑腸溶口崩片����。微丸壓片在初期考察時出現(xiàn)黏沖現(xiàn)象,可能是由于口崩片中含有大量的甘露醇�����、疏水性的MCC用量較少導(dǎo)致黏沖�,此外,硬脂酸鎂用量也會影響?zhàn)_情況�。壓片過程中,如果腸溶微丸在片中的占比過多�,微丸在壓片的時候被壓碎,會導(dǎo)致在酸中的溶出出現(xiàn)突釋現(xiàn)象����。口崩片的崩解時限受崩解劑和MCC的影響����。
本品為腸溶制劑,在體外釋放度方法開發(fā)過程中���,選用了不同pH的釋放介質(zhì)����,模擬藥物進(jìn)入人體消化道后的過程,并對蘭索拉唑在各所用釋放介質(zhì)中的紫外吸收進(jìn)行了考察��,結(jié)果見圖3�。蘭索拉唑在溶出介質(zhì)Ⅰ和Ⅱ中的最大吸收波長分別為332和286nm,因此確定了體外釋放度的檢測波長�。
本研究以崩解時限�、黏沖情況、脆碎度和溶出度作為指標(biāo)��,通過四因素三水平的正交實驗���,篩選出了優(yōu)化后的處方�,并用優(yōu)化后的處方進(jìn)行連續(xù)3批的樣品制備�����。結(jié)果表明�����,以優(yōu)化后的處方制備的樣品溶出度��、崩解時限���、脆碎度����、含量和含量均勻度均符合要求。穩(wěn)定性考察結(jié)果表明�,40℃放置6個月樣品脆碎度、崩解時限��、含量���、有關(guān)物質(zhì)和溶出度均無明顯變化���,產(chǎn)品穩(wěn)定性良好。