摘要:該文是在國(guó)家鼓勵(lì)高質(zhì)量推動(dòng)中藥產(chǎn)品國(guó)際注冊(cè)的大背景下�,針對(duì)美國(guó)植物藥注冊(cè)申請(qǐng)的藥學(xué)要求開展的研究。研究以現(xiàn)行版《美國(guó)植物藥開發(fā)工業(yè)指南》為基礎(chǔ)�����,以我國(guó)在FDA 注冊(cè)申報(bào)的復(fù)方丹參滴丸和連花清瘟膠囊2個(gè)產(chǎn)品以及USFDA 已批準(zhǔn)的4 個(gè)植物藥處方藥產(chǎn)品為案例���,基于公開信息剖析其中的藥學(xué)研究?jī)?nèi)容���,探究美國(guó)植物藥注冊(cè)申請(qǐng)的藥學(xué)研究關(guān)注點(diǎn)和難點(diǎn),以期為我國(guó)中藥1.2 類提取物及其制劑�、天然藥物的研發(fā)和我國(guó)中藥在FDA 以植物藥處方藥注冊(cè)申請(qǐng)?zhí)峁﹨⒖己徒梃b。
美國(guó)植物藥由植物組成�����,包括植物材料���、藻類���、大型真菌,或者上述物質(zhì)的組合���。劑型可以是溶液劑(例如茶劑)���、粉劑、片劑�����、膠囊、糖漿�、外用制劑或注射劑。發(fā)酵類���、高純度���、化學(xué)修飾的植物原材料等不屬于植物藥范疇。目前美國(guó)已批準(zhǔn)4 個(gè)滿足植物藥指南定義的新植物藥處方藥[1]�。
1 植物藥的特點(diǎn)及其所面臨的科學(xué)和注冊(cè)方面的挑戰(zhàn)
植物藥源自天然,由復(fù)雜的混合物組成�����,活性成份不明確�����,多種化學(xué)成分可能對(duì)生理或藥理作用有貢獻(xiàn)�,其化學(xué)成分及生物活性,在質(zhì)量控制方面通常無法得到很好的表征�����。此外�����,植物藥還存在較大的批間變異性,這種變異性比化學(xué)藥的批間差異更為顯著�����?��;瘜W(xué)藥的常規(guī)質(zhì)量控制方法對(duì)于植物藥來說也是不充分的,植物藥通常需采用多種方法進(jìn)行聯(lián)合檢測(cè)���。相比于化學(xué)藥�,植物藥通常具有先前的人用經(jīng)驗(yàn)���,可以為早期臨床試驗(yàn)安全性提供一些提示�����,但在后期臨床試驗(yàn)中���,需評(píng)價(jià)批間差異對(duì)植物藥療效的影響。
在管理方面�,植物藥與其他藥品一樣,均需符合同樣的法律標(biāo)準(zhǔn)[2]。但是植物藥提供的某些資料可以與新化學(xué)實(shí)體(NCE)不同�,主要包括以下方面,見表1�����。

由于植物藥的上述特點(diǎn)�����,使得對(duì)植物藥來源�、生產(chǎn)工藝步驟、標(biāo)準(zhǔn)化和處方過程等方面的理解���,對(duì)植物藥制劑的特性鑒定和一致性評(píng)價(jià)都至關(guān)重要���。此外,植物藥的生物作用機(jī)制可能未知或由于過于復(fù)雜而無法明確獲知�。當(dāng)無法進(jìn)行藥理學(xué)和藥代動(dòng)力學(xué)試驗(yàn)時(shí),可以豁免�����,但在缺乏此類數(shù)據(jù)的情況下���,可能更難確定植物藥的“洗脫”期�����,或預(yù)測(cè)和研究“藥物-藥物”相互作用���。植物藥在臨床應(yīng)用中的很多處理方式與復(fù)雜的生物制劑更為相似�����,美國(guó)食品藥品監(jiān)督管理局(USFDA)采用的處理方法更類似于生物制劑��。由于植物藥的特性無法全面表征���,因此,植物藥通常通過過程控制和驗(yàn)證來定義���,即“過程即產(chǎn)品�����,產(chǎn)品即過程”�����。不過���,這種復(fù)雜性反而使得植物藥在沒有專利保護(hù)的情況下也能得到知識(shí)產(chǎn)權(quán)保護(hù)�����。在審評(píng)植物藥申請(qǐng)時(shí)���,USFDA 會(huì)考慮這些因素,并根據(jù)“個(gè)案處理”的原則向申辦方提供建議[3]���。對(duì)于以植物原材料為起始物料的藥品���,其生產(chǎn)首先要確保植物原材料(botanical rawmaterial,BRM)的鑒定���,此外�����,應(yīng)該考慮BRM 的可及性及持續(xù)供應(yīng)���,上述內(nèi)容都屬于良好種植和采收管理規(guī)范(GACP)的范疇��。USFDA 通常把植物原料藥(botanical drug substance�����,BDS)整體作為活性成分�����,BDS 的表征較難�����,可以通過生物標(biāo)志物和化學(xué)指紋進(jìn)行表征;每個(gè)工藝步驟均應(yīng)進(jìn)行詳細(xì)的描述���;全過程的控制有助于確保臨床上所用劑量的可重復(fù)性以及批間一致性���。
2 美國(guó)植物藥的藥學(xué)研究要求
基于植物藥的上述特點(diǎn)和注冊(cè)方面的挑戰(zhàn),USFDA 專門起草了《美國(guó)植物藥開發(fā)工業(yè)指南》用于指導(dǎo)植物藥的研發(fā)和注冊(cè)申報(bào)[4]�����。本文將集中在其中的藥學(xué)內(nèi)容進(jìn)行概括��、梳理和探究。
2.1 不同研究階段的一些總體要求和建議
2.1.1 在通用技術(shù)要求方面���,植物藥的與其他藥品一致
植物藥是藥品的一種�����,在質(zhì)量控制和臨床試驗(yàn)設(shè)計(jì)方面與其他藥品相比有其特殊性��,但是同時(shí)它也需要遵循其他藥品的一些通用要求��。例如:植物藥制劑的新藥申請(qǐng)需要采用電子通用技術(shù)文檔(eCTD)格式遞交���;穩(wěn)定性試驗(yàn)參考ICHQ1A(R2)的科學(xué)原理進(jìn)行穩(wěn)定性試驗(yàn)設(shè)計(jì);植物原料藥可以作為臨床試驗(yàn)申請(qǐng)(IND)��、上市許可申請(qǐng)(NDA)資料的一部分遞交��,也可以以藥物主文件(DMF)的形式遞交��。
2.1.2 注冊(cè)資料的提交要求
植物藥的臨床開發(fā)采取循序漸進(jìn)的方式�����,以便于采用早期(I期��、II期)的臨床試驗(yàn)結(jié)果支持后期(III 期)的臨床試驗(yàn)設(shè)計(jì)。早期臨床試驗(yàn)需提交的資料取決于以下因素:先前的人用經(jīng)驗(yàn)和既往臨床試驗(yàn)的研究程度�����、藥物的已知或可能風(fēng)險(xiǎn)以及藥物的開發(fā)階段��。對(duì)大多數(shù)植物藥來說�����,早期臨床試驗(yàn)階段可能無需提供詳細(xì)的藥學(xué)(CMC)資料(例如:原料的全面表征數(shù)據(jù))�����,在III期臨床試驗(yàn)啟動(dòng)前需要提交較完善的藥學(xué)研究資料��。
2.1.3 留樣建議
在早期臨床試驗(yàn)時(shí)���,臨床試驗(yàn)用樣品應(yīng)留樣,以備USFDA 和/或申辦方未來的檢驗(yàn)���。臨床樣品所用的植物原材料和原料藥生產(chǎn)商的每一批樣品均應(yīng)留樣�����;在???期臨床試驗(yàn)時(shí)��,應(yīng)保留充足的�����、不同批次的植物原材料和原料藥��,以備后續(xù)的化學(xué)表征和/或藥理/毒理試驗(yàn)���。
2.1.4 穩(wěn)定性試驗(yàn)要求
在早期臨床試驗(yàn)時(shí)�����,原料藥需開發(fā)穩(wěn)定性指示性的分析方法�����,制劑穩(wěn)定性數(shù)據(jù)應(yīng)至少支持臨床試驗(yàn)期間的使用��。穩(wěn)定性試驗(yàn)貫穿藥品上市的全過程���,并最終支持?jǐn)M定的有效期。在新藥申請(qǐng)時(shí),穩(wěn)定性試驗(yàn)方案中應(yīng)包含生物測(cè)定方法���,申請(qǐng)人應(yīng)開發(fā)��、驗(yàn)證和使用具有穩(wěn)定性指示性的分析方法和/或生物測(cè)定方法���,以監(jiān)測(cè)植物原料藥和制劑的穩(wěn)定性。申請(qǐng)人還應(yīng)進(jìn)行影響因素穩(wěn)定性試驗(yàn)�����,以識(shí)別原料藥和制劑的降解產(chǎn)物���,評(píng)估潛在的毒性���,并對(duì)這些降解產(chǎn)物進(jìn)行充分控制。
2.2 不同研究階段�����,針對(duì)可能的變化��,需要開展的橋接研究
2.2.1 早期臨床試驗(yàn)
由于植物原材料在種植和采收過程中可能的變化和/或工藝優(yōu)化而導(dǎo)致的生產(chǎn)工藝條件的變化��,進(jìn)而導(dǎo)致不同開發(fā)階段所用的植物原料藥可能會(huì)在某些特性上有所不同(例如化學(xué)組成)���,需要通過橋接研究來說明這些變化對(duì)質(zhì)量產(chǎn)生的影響��,橋接研究的類型需要與新藥辦公室(OND)的審評(píng)部門溝通�����。
2.2.2 III期臨床試驗(yàn)
在III期臨床試驗(yàn)前�����,原料藥和制劑的藥學(xué)開發(fā)應(yīng)足夠深入��,以確保在III期臨床試驗(yàn)過程中不對(duì)植物原材料和生產(chǎn)工藝進(jìn)行重大變更���,從而確保臨床試驗(yàn)用原料藥與擬用于上市的原料藥一致。
如果 IND 階段提供了或引用了早期開發(fā)階段的非臨床和/或臨床數(shù)據(jù)��,申辦方應(yīng)提供IND 所用植物藥制劑與引用研究中所用植物藥制劑間的比較研究(例如:原料藥中活性成分或化學(xué)成分的特性鑒定和定量測(cè)定��、制劑的組成和配方等)��。同樣��,在開發(fā)過程中,當(dāng)植物原材料���、原料藥或制劑的來源和生產(chǎn)工藝發(fā)生變更時(shí)���,申請(qǐng)人應(yīng)對(duì)變更前后進(jìn)行對(duì)比,來源和/或生產(chǎn)工藝看似微小的變更�����,也可能導(dǎo)致臨床療效的顯著性差異�����,進(jìn)而會(huì)導(dǎo)致早期的藥理�����、非臨床和臨床數(shù)據(jù)不可用�����。如果無法確定不同批次原料藥是否相似��,應(yīng)提供橋接研究(例如:原料藥中活性或化學(xué)成分的化學(xué)鑒定和定量分析��、生物測(cè)定�����、和/或其他非臨床研究)來證明不同開發(fā)階段原料藥的充分相似性���,進(jìn)而判定先前的非臨床研究和臨床試驗(yàn)結(jié)果是否可用��。
處方的變更若導(dǎo)致先前開展的藥理或毒理研究無法適用于新的處方��,可能需要提交非臨床橋接研究���。申辦方應(yīng)就擬定的處方變更與審評(píng)部門進(jìn)行咨詢,確定變更是否需要橋接研究或其他研究��。值得注意的是���,如果在臨床試驗(yàn)開發(fā)階段��,原料藥和制劑發(fā)生變更��,可能需要提供非臨床橋接研究���。
2.2.3 新藥申請(qǐng)階段
后期臨床試驗(yàn)用的原材料的采收�����、加工方法應(yīng)與早期所用方法相同��。臨床開發(fā)過程所用的原材料的采收���、加工方法發(fā)生變更的,可能會(huì)改變?cè)纤?�、制劑的化學(xué)組成�����,需要提交橋接研究以證明先前的臨床試驗(yàn)結(jié)果仍然適用�����。
2.2.4 上市后階段
植物藥制劑變更所產(chǎn)生的影響可能不易識(shí)別���,有必要開展額外的研究(例如:生物測(cè)定和/或其他體內(nèi)橋接研究)��,評(píng)估效力和活性��。USFDA 將根據(jù)變化的性質(zhì)和程度以及混合物中活性成分或化學(xué)成分表征的程度等具體情況�����,要求申辦方提供相應(yīng)的研究數(shù)據(jù)��。
2.3 生物測(cè)定法
生物測(cè)定法是一種測(cè)定植物藥效力和活性的重要方法�����。首選與藥物已知或預(yù)期作用機(jī)制相關(guān)的生物測(cè)定法��。某些情況下���,可以考慮采用相關(guān)性較低的檢測(cè)方法進(jìn)行評(píng)估。由于生物測(cè)定結(jié)果的變異性較大��,所以應(yīng)測(cè)定相對(duì)于對(duì)照品或?qū)φ瘴镔|(zhì)的批效力和活性�����,生物測(cè)定結(jié)果應(yīng)以對(duì)照品或?qū)φ瘴镔|(zhì)校正的活性單位來表示��。申請(qǐng)人應(yīng)開展系統(tǒng)適用性考察和質(zhì)量控制研究�����,以確保檢驗(yàn)方法的重復(fù)性和可預(yù)測(cè)性。如果植物藥的活性成分未知或無法定量���,在III期臨床試驗(yàn)開始前���,應(yīng)開發(fā)生物測(cè)定方法,以評(píng)價(jià)原料藥相對(duì)于對(duì)照物質(zhì)的批效力和活性���;在提交新藥申請(qǐng)時(shí)�����,申請(qǐng)人應(yīng)對(duì)生物測(cè)定法進(jìn)行適當(dāng)?shù)尿?yàn)證�����,驗(yàn)證指標(biāo)至少應(yīng)包括準(zhǔn)確性��、精密度��、專屬性�����、線性和范圍�����。如果質(zhì)量控制需要采用生物測(cè)定法��,質(zhì)量平衡應(yīng)與生物測(cè)定法聯(lián)合進(jìn)行評(píng)價(jià)��。
穩(wěn)定性試驗(yàn)方案中應(yīng)該包含生物測(cè)定法��,變更研究時(shí)生物測(cè)定結(jié)果也是一項(xiàng)關(guān)鍵的對(duì)比指標(biāo)��。
2.4 質(zhì)量平衡
在III期臨床試驗(yàn)開始前���,應(yīng)開發(fā)其他(例如脂質(zhì)或蛋白質(zhì))定量方法,以測(cè)定對(duì)原料藥質(zhì)量平衡有貢獻(xiàn)的其他類成份�����。
在新藥申請(qǐng)階段�����,植物藥開發(fā)過程中積累的經(jīng)驗(yàn)和數(shù)據(jù)應(yīng)為制定臨床相關(guān)標(biāo)準(zhǔn)奠定基礎(chǔ)���,分析方法應(yīng)通過適當(dāng)驗(yàn)證���。此外��,當(dāng)聯(lián)合使用多種方法時(shí)�����,所有分析方法所得到的整體數(shù)據(jù)��,應(yīng)可以證明檢驗(yàn)樣品中不同類成分間和同一類成份內(nèi)(如果恰當(dāng))的質(zhì)量平衡��。分析方法應(yīng)足夠敏感���,以檢測(cè)多批次關(guān)鍵質(zhì)量屬性之間的差異。如果可能��,原料藥應(yīng)包括以下檢驗(yàn)內(nèi)容:各已知化學(xué)成分的重量和同類成分的總量���、各未知峰面積百分比���、各未知可量化峰的相對(duì)保留時(shí)間(RRT)、總脂和各脂肪酸的重量���、總氨基酸和各氨基酸的重量�����、簡(jiǎn)單碳水化合物總重量��、復(fù)雜碳水化合物總重量���、總維生素和各維生素重量��、灰分含量等��。
2.5 確保植物藥療效一致性需要提供的證據(jù)
植物藥制劑生產(chǎn)商面臨的重大挑戰(zhàn)之一是確保不同上市制劑批次(盡管有變異性)的療效與III期臨床試驗(yàn)用樣品療效的一致性��。由于植物藥制劑天然的變異性,僅靠化學(xué)檢測(cè)可能不足以控制其質(zhì)量��,無法保障療效的一致性�����。植物藥制劑的質(zhì)量控制應(yīng)從以下3 個(gè)方面考慮:①植物原材料的控制���,②化學(xué)檢測(cè)的質(zhì)量控制和生產(chǎn)控制�����,③生物測(cè)定和臨床數(shù)據(jù)��。申請(qǐng)人應(yīng)對(duì)上述3 個(gè)質(zhì)量控制方面進(jìn)行綜合評(píng)估�����,以確定所需的數(shù)據(jù)量�����,例如���,①和/或③(生物測(cè)定)所需的數(shù)據(jù)量可能取決于②化學(xué)檢測(cè)對(duì)植物混合物中成分表征的程度��,從而證明上市后的各制劑批次與上市前臨床試驗(yàn)用樣品療效一致�����。NDA 申請(qǐng)時(shí)�����,這部分資料放在模塊2.3.P.2(藥學(xué)開發(fā))項(xiàng)下植物藥特有的章節(jié)“治療一致性保證”��。
盡管原料藥控制和其他 CMC 控制措施有助于產(chǎn)品的特性鑒定���,保證質(zhì)量�����,但在某些情況下��,可能需要提供有關(guān)質(zhì)量指標(biāo)與藥理學(xué)活性或臨床療效之間的相關(guān)性���,以確保原材料和原料藥的變化不會(huì)影響制劑的療效一致性。臨床數(shù)據(jù)包括劑量—效應(yīng)數(shù)據(jù)和多批次制劑的數(shù)據(jù)�����。通過劑量—效應(yīng)數(shù)據(jù)��,若發(fā)現(xiàn)臨床療效對(duì)劑量不敏感(但仍優(yōu)于安慰劑對(duì)照組)�����,則有理由推測(cè)�����,質(zhì)量標(biāo)準(zhǔn)范圍內(nèi)的變化可能不會(huì)影響藥品的療效一致性��;多批次制劑的臨床數(shù)據(jù)���,是指臨床試驗(yàn)中將受試者隨機(jī)分配到不同的制劑批次���,用于評(píng)價(jià)療效與制劑批次的相互作用。若相互作用不明顯�����,說明質(zhì)量標(biāo)準(zhǔn)范圍內(nèi)藥物批次的變化不會(huì)影響療效[5]�����。
2.6 不同研究階段對(duì)植物原材料(BRM)���、植物原料藥(BDS)和植物藥制劑(BDP)的具體要求
2.6.1 早期臨床試驗(yàn)階段
2.6.1.1 BRM
應(yīng)該提供先前人用經(jīng)驗(yàn)中有關(guān)BRM 的以下信息:國(guó)際雙名法慣例命名���、異名、種(亞種��、變種和變型)和栽培變種�����、科名、藥用部位���;關(guān)于化學(xué)組成�����,應(yīng)識(shí)別已知活性成分群(確定為特定化合物群或化學(xué)類別)和未知活性成分群的化學(xué)成分(例如���,作為鑒別和質(zhì)量控制的特征)。
支持 IND 所需的CMC 信息量取決于很多因素���,包括植物藥的上市歷史和研發(fā)階段��,在美國(guó)境外上市的植物藥人用經(jīng)驗(yàn)數(shù)據(jù)要求取決于境外生產(chǎn)該植物藥的控制措施和質(zhì)量控制的完整性�����。
對(duì) BRM 的控制包括通過感官��、外觀和顯微方法鑒別植物基原、藥用部位等���,以及通過與憑證標(biāo)本(即對(duì)照標(biāo)本)對(duì)比進(jìn)行的鑒別研究��,還可以通過遺傳分類學(xué)的方法進(jìn)行鑒別(DNA 條形碼技術(shù))��。如果所用物種包含多個(gè)變種時(shí)���,應(yīng)明確每個(gè)變種���。植物原材料和原料藥生產(chǎn)商的每一批次樣品均應(yīng)留樣,并在適當(dāng)?shù)臈l件下保存植物及其藥用部位或其他植物材料��。必要時(shí)�����,可以用這些樣品來鑒別真?zhèn)巍?/span>
需要提供的資料包括:植物種及其藥用部位的鑒定�����、真品憑證���、物種是否屬于瀕危物種及是否生長(zhǎng)在瀕臨滅絕或受到威脅的產(chǎn)地中���。
應(yīng)提供每個(gè)種植者和/或供應(yīng)商(如果有)的名稱和地址�����,植物種(變種和栽培變種)的外觀和特征���,以及植物鑒定(外觀和顯微)信息,產(chǎn)地(全球定位系統(tǒng)GPS 坐標(biāo))��、生長(zhǎng)條件��、采收時(shí)植物的生長(zhǎng)階段�����、采收時(shí)間/季節(jié)��,和后續(xù)的加工處理(例如:清洗�����、干燥��、研磨工序)���;雜質(zhì)控制(即:無機(jī)和有機(jī)污染物��,如土壤�����、昆蟲和藻類/真菌)�����;保存��;搬運(yùn)�����、運(yùn)輸和貯藏條件���;元素雜質(zhì)檢測(cè);微生物限度���;殘留農(nóng)藥檢測(cè)�����;和外源性毒素檢測(cè)(例如黃曲霉毒素)��、外來物質(zhì)與混偽品等�����。
2.6.1.2 BDS
應(yīng)提供原料藥的定性描述�����、定量描述�����、生產(chǎn)商�����、生產(chǎn)工藝描述���、質(zhì)量控制信息等�����,具體內(nèi)容如下���。
原料藥的定性描述包括名稱�����、外觀���、活性成分�����、理化性質(zhì)��、生物活性�����、以及先前臨床中用于制備原料藥的各個(gè)植物原材料�����。如果活性成份�����、生物活性或先前臨床使用情況未知,應(yīng)該在申請(qǐng)時(shí)闡明��。對(duì)于多種植物組成的原料藥�����,申請(qǐng)時(shí)應(yīng)說明原料藥的各植物原材料單獨(dú)加工成原料藥后再混合還是混合后再加工�����。
原料藥的定量描述應(yīng)包括:植物原材料的規(guī)格(一般以加工后的植物原材料的絕對(duì)干重計(jì))��;應(yīng)該提供批量以及相對(duì)于植物原材料的得率��;當(dāng)活性成分或其他化學(xué)成份已知���、可測(cè)時(shí)��,應(yīng)該明確其在植物原材料中的含量��;對(duì)于多種植物組成的原料藥��,應(yīng)該闡述每個(gè)加工后植物原料藥或加工前植物原材料(若適用)的相對(duì)比例���。
生產(chǎn)工藝描述�����。應(yīng)包括每一個(gè)工序藥材投料量���、溶劑、提取和/或干燥的溫度和時(shí)間及生產(chǎn)過程控制�����;應(yīng)標(biāo)明加工收率��,以原藥材的量相對(duì)于提取物的量來表示�����;如果用多種植物原材料混合生產(chǎn)含有多種植物的一個(gè)原料藥�����,應(yīng)提供每個(gè)藥材的投料量及其加入順序��、混合���、研磨���、和/或提取等信息;如果多植物原料藥是由兩種或兩種以上單獨(dú)制備的植物原料藥共同組成�����,應(yīng)對(duì)每個(gè)原料藥的工藝進(jìn)行單獨(dú)描述�����。
原料藥的質(zhì)量控制��。對(duì)每批原料藥進(jìn)行質(zhì)量控制�����,包括所用的分析方法�����、檢驗(yàn)結(jié)果及擬定的可接受標(biāo)準(zhǔn)���。質(zhì)量控制檢驗(yàn)項(xiàng)目至少應(yīng)該包括:外觀���、以干重計(jì)的規(guī)格(相當(dāng)于植物原材料的量)��、活性成份(如果已知)或化學(xué)成份的鑒別和含量測(cè)定���。一般來說,申辦方應(yīng)該采用可用的分析技術(shù)解決分析方法的分離度問題���。應(yīng)采用多種方法互相補(bǔ)充確保對(duì)成份進(jìn)行充分的化學(xué)定性和定量表征���。若采用多種植物原材料混合生產(chǎn)一個(gè)原料藥,無法對(duì)每個(gè)活性成分或化學(xué)成分進(jìn)行定量測(cè)定時(shí)��,可以建立一種聯(lián)合方法對(duì)多個(gè)活性或化學(xué)成分群進(jìn)行測(cè)定��。當(dāng)多個(gè)活性成分或化學(xué)成分已知時(shí)��,應(yīng)對(duì)其進(jìn)行化學(xué)表征���,并對(duì)其相對(duì)含量進(jìn)行定義。此外��,還應(yīng)對(duì)農(nóng)藥殘留(經(jīng)常使用的農(nóng)藥)���、無機(jī)雜質(zhì)�����、殘留溶劑和放射性污染(如果有的話)��、微生物限度��、外源性毒素(如黃曲霉毒素)等進(jìn)行檢驗(yàn)�����,以及開展穩(wěn)定性試驗(yàn)�����、生物測(cè)定法以及質(zhì)量平衡等研究�����。
2.6.1.3 BDP
植物藥制劑應(yīng)提供以下資料:①制劑的定性描述��,如劑型�����、給藥途徑��、組成制劑的物料名稱和功能(例如:植物原料藥�����、其他原料藥和輔料)��,對(duì)于植物原料藥與其他原料藥(例如���,高純度的�����、生物技術(shù)的��,或其他天然來源的藥物原料)的復(fù)方��,應(yīng)予以說明���;②制劑的組成或定量描述���,以單位劑量和批處方量來表示���;③制劑生產(chǎn)商的檢驗(yàn)報(bào)告或制劑生產(chǎn)商授權(quán)USFDA 對(duì)生產(chǎn)商之前提交的資料或DMF 中的相關(guān)CMC 信息進(jìn)行交叉引用�����。如果國(guó)外上市產(chǎn)品沒有這部分信息�����,則申辦方應(yīng)對(duì)制劑進(jìn)行質(zhì)量檢驗(yàn)��。除了這些檢驗(yàn)外�����,應(yīng)開展無機(jī)雜質(zhì)分析和動(dòng)物安全性試驗(yàn)(如適用)���,申辦方應(yīng)當(dāng)在IND 時(shí)提供檢驗(yàn)方法和結(jié)果。留樣建議以及穩(wěn)定性數(shù)據(jù)要求在前面已有闡述��。
特殊情況下�����,如原料藥的活性成分已知�����,且藥材中這些活性成分的濃度有較大的、天然的變化(例如:因生長(zhǎng)條件隨時(shí)間推移而產(chǎn)生的難以控制的變化)��,允許植物藥制劑增加單體活性成分的含量以滿足基準(zhǔn)原料藥的質(zhì)量標(biāo)準(zhǔn)�����,以及療效的批間一致性���。但加入的活性成分目標(biāo)含量不應(yīng)超過天然含量。關(guān)于加入活性成分含量的恰當(dāng)與否以及確定基準(zhǔn)原料藥標(biāo)準(zhǔn)的過程等相關(guān)事宜�����,申辦方應(yīng)事先向USFDA 咨詢���。
安慰劑��。必要時(shí)�����,可在安慰劑中使用某些植物材料來掩蓋活性藥物��,但這類植物材料不應(yīng)有明確的藥理活性�����,如有活性�����,將使臨床試驗(yàn)數(shù)據(jù)難以解釋��。對(duì)于一些植物藥來說���,很難制備出一種在味道、氣味和外觀上與活性藥物相同的安慰劑�����。如果研究者和受試者區(qū)分不出活性藥物與安慰劑��,并且可以保證臨床試驗(yàn)的盲法���,那么即便安慰劑與試驗(yàn)藥物間有細(xì)微差別也是可以接受的�����。就該類安慰劑的使用�����,鼓勵(lì)申請(qǐng)人向OND 相應(yīng)的審評(píng)部門咨詢�����。
2.6.2 III期臨床試驗(yàn)階段
植物藥深入而持續(xù)的表征可以確保原料藥的質(zhì)量���,進(jìn)而保障臨床數(shù)據(jù)的有效性和可靠性��。
不同批次植物原料藥間會(huì)有批間差異(例如:化學(xué)成分的變化)��,申辦方應(yīng)提供證據(jù)證明原料藥的批間差異不會(huì)對(duì)療效產(chǎn)生明顯影響���。其中的一種方法是在III期臨床試驗(yàn)中使用多批次的植物藥制劑(即每批制劑采用不同批次的植物原材料生產(chǎn)),以研究不同批次制劑間的臨床療效���。這些研究將有利于申辦方更好地了解與臨床療效相關(guān)的變化因素���,以及在多大范圍內(nèi)的變異仍可保證植物藥制劑的質(zhì)量、有效性和安全性��。這種深入的研究將有助于制定臨床相關(guān)質(zhì)量標(biāo)準(zhǔn)的可接受限度。
2.6.2.1 BRM
為評(píng)價(jià)質(zhì)量和療效的一致性��,選擇具有代表性的BRM 批次生產(chǎn)臨床試驗(yàn)用原料藥�����,進(jìn)而生產(chǎn)多批???期臨床試驗(yàn)用制劑��。申請(qǐng)人應(yīng)建立3 個(gè)或更多的種植基地或農(nóng)場(chǎng)等大種植區(qū)��,這些種植區(qū)應(yīng)選擇在各BRM 的代表性區(qū)域中���,并按照GACP 的原則進(jìn)行種植。這將有助于在NDA 批準(zhǔn)后減少藥材資源不足的風(fēng)險(xiǎn)�����。
申請(qǐng)人應(yīng)提供藥材表征的進(jìn)一步研究信息(例如:通過光譜或色譜法對(duì)每個(gè)藥材進(jìn)行化學(xué)鑒定��,必要時(shí)用DNA 指紋圖譜法對(duì)每個(gè)藥材進(jìn)行真?zhèn)舞b定)��,更新藥材供應(yīng)商所用的質(zhì)量控制方法��、分析方法以及擬定的質(zhì)量標(biāo)準(zhǔn)�����,以便為NDA 資料遞交做準(zhǔn)備。
2.6.2.2 BDS 及BDP
申請(qǐng)人應(yīng)建立相關(guān)質(zhì)量標(biāo)準(zhǔn)(含暫定的可接受標(biāo)準(zhǔn))���,并根據(jù)???期臨床試驗(yàn)結(jié)果在NDA 申報(bào)時(shí)確定最終的質(zhì)量標(biāo)準(zhǔn)��。
2.6.3 新藥申請(qǐng)階段
鑒于植物藥制劑的獨(dú)特性和特殊考慮��,新藥申請(qǐng)前會(huì)議尤其重要���。一般來說,植物藥制劑的NDA 資料提交要求與其他藥物相同��。
在 IND 階段提交的有關(guān)制劑描述和先前人用經(jīng)驗(yàn)資料的所有信息都應(yīng)該在NDA 中提交�����,若有最新的人用經(jīng)驗(yàn)資料(例如:基于已在國(guó)外銷售的類似產(chǎn)品)��,應(yīng)進(jìn)行更新�����。
質(zhì)量控制���。由于原料藥的活性成分可能未被識(shí)別�����,質(zhì)量控制的技術(shù)挑戰(zhàn)是確定植物藥的特性并確保其療效的一致性���。植物藥制劑質(zhì)量控制的整體證據(jù)鏈條法(totality-of-the-evidence approach)應(yīng)延伸到對(duì)原材料可能采取的額外的控制措施��,如生物測(cè)定和/或多批次臨床試驗(yàn)中BRM 質(zhì)量差異對(duì)臨床結(jié)局的影響。對(duì)植物藥的鑒定不僅在于對(duì)混合物中化學(xué)成份的特性鑒定�����,還要依賴化學(xué)檢測(cè)的質(zhì)量控制和生產(chǎn)控制��、生物測(cè)定���、臨床數(shù)據(jù)等�����。
2.6.3.1 BRM
植物制劑的質(zhì)量控制應(yīng)從 BRM 開始�����,并在NDA 中進(jìn)行闡述��。應(yīng)明確藥用植物(如采用形態(tài)學(xué)��、宏觀和微觀分析���、化學(xué)分析等方法鑒定真?zhèn)危?��、種植規(guī)范(如生長(zhǎng)、采收時(shí)間���、貯存條件)��、地理位置�����,以及采收和加工方法的具體信息�����。申請(qǐng)人在提交NDA 時(shí)���,應(yīng)建立了GACP 基地�����,并匯總每種BRM 的相關(guān)規(guī)程���。應(yīng)參照世界衛(wèi)生組織(WHO)、歐洲藥品管理局(EMA)或BRM 種植區(qū)監(jiān)管機(jī)構(gòu)制定的GACP 通則���。當(dāng)植物的分類較復(fù)雜��,或存在BRM 鑒定問題時(shí)���,可以采用DNA 指紋圖譜鑒定���。例如��,如果多個(gè)相關(guān)植物藥種屬用于生產(chǎn)某特定藥材時(shí)��,DNA 指紋鑒別方法較其他鑒別方法專屬性更強(qiáng)���。此外,申請(qǐng)人應(yīng)描述減少植物污染��、退化和變異的方法,早期和后期臨床試驗(yàn)所用的植物原材料應(yīng)該使用同樣的方法���。在臨床開發(fā)過程中���,這些采收加工方法上的變化可能會(huì)改變?cè)纤帲M(jìn)而導(dǎo)致植物藥制劑的化學(xué)組成的變化���,需要提交橋接研究以證實(shí)先前臨床試驗(yàn)結(jié)果的可靠性���。
2.6.3.2 BDS 和BDP
①特性鑒定。表征植物藥的所有化學(xué)成分存在一定困難�����,不僅體現(xiàn)在對(duì)混合物的化學(xué)特性進(jìn)行表征��,還包括對(duì)植物原材料的控制���、生產(chǎn)控制��、生物測(cè)定�����、臨床數(shù)據(jù)等�����。即便如此��,申請(qǐng)人還應(yīng)利用現(xiàn)有技術(shù)和新技術(shù)開發(fā)正交分析法��,對(duì)植物藥中活性成份或化學(xué)成分進(jìn)行充分定性和定量�����。當(dāng)活性成分未知��、植物混合物不能被完全表征時(shí)��,申請(qǐng)人可以選擇一種化學(xué)成分特征譜(這種特征譜對(duì)BRM 質(zhì)量和/或原料藥���、制劑生產(chǎn)條件變化較敏感)進(jìn)行特性鑒定。②化學(xué)特征���。在NDA 階段應(yīng)充分闡述用于植物藥化學(xué)成份表征的多種分析方法�����。NDA 階段應(yīng)包含用于定性和定量表征的活性成份或化學(xué)成分的所有化學(xué)檢測(cè)方法��,也包括提供數(shù)據(jù)說明質(zhì)量平衡��。③生產(chǎn)工藝��。申請(qǐng)人應(yīng)提供用于商業(yè)化生產(chǎn)原料藥和制劑生產(chǎn)場(chǎng)地的所有信息�����。應(yīng)避免變更原料藥的生產(chǎn)場(chǎng)地�����,特別是臨床開發(fā)后期�����。NDA 應(yīng)提供完整的生產(chǎn)信息���,包括所用的生產(chǎn)設(shè)備���、中間控制和檢驗(yàn)。④其他。還應(yīng)提供生物測(cè)定法���、質(zhì)量平衡等質(zhì)量控制資料以及穩(wěn)定性試驗(yàn)資料��。
2.6.4 cGMP 要求
鑒于植物藥活性成分的異質(zhì)性和不確定性��,對(duì)植物原材料的控制�����,包括貯存條件和加工方法�����,都極其重要�����。申請(qǐng)人應(yīng)在申請(qǐng)中提供充分的起始物料質(zhì)量信息���,而不僅僅依靠對(duì)終產(chǎn)品的質(zhì)量控制。植物原料藥的生產(chǎn)應(yīng)符合cGMPs���。在某些情況下,可能需要同時(shí)符合GACP 和cGMPs,以覆蓋藥材的種植��、采收�����、加工和貯存的各個(gè)方面���。申請(qǐng)人應(yīng)指定原料藥生產(chǎn)的起點(diǎn)并論證其確定依據(jù)��。
2.7 上市后考慮
鑒于 BRM 穩(wěn)定來源和原料藥生產(chǎn)工藝一致性的重要性��,制劑批準(zhǔn)后若對(duì)產(chǎn)地���、種植和采收規(guī)范、和/或加工方法等進(jìn)行變更���,均須仔細(xì)評(píng)估��,以確保變更前后所生產(chǎn)的制劑在藥理和/或療效上的充分相似性���。對(duì)于植物藥制劑,變更的影響可能不易評(píng)價(jià)��,必要時(shí)需開展額外研究(例如采用生物測(cè)定和/或其他體內(nèi)橋接研究)來評(píng)估其效力和活性。針對(duì)變更所需要的研究數(shù)據(jù)���,F(xiàn)DA 會(huì)具體問題具體分析���,例如會(huì)考慮變更的性質(zhì)和程度以及混合物中活性成分或化學(xué)成分的變化程度。
3 我國(guó)中藥在USFDA 申報(bào)品種的藥學(xué)研究情況
根據(jù)文獻(xiàn)報(bào)道��,我國(guó)已有一些中藥品種在美國(guó)獲批了IND 申請(qǐng)���,主要開展了II期臨床試驗(yàn)��,進(jìn)入III期的寥寥無幾��?�;诳刹樵兊降墓_文獻(xiàn)信息�����,以2 個(gè)品種作為案例���,介紹其藥學(xué)研究情況:
3.1 復(fù)方丹參滴丸(T89)
復(fù)方丹參滴丸于 1997 年以天然復(fù)方混合制劑的形式首次申報(bào)USFDA 的IND 申請(qǐng),直接進(jìn)入新藥II~I(xiàn)I期臨床試驗(yàn)��,2006 年再次通過USFDA 的IND 申請(qǐng)���,2008 年在美國(guó)啟動(dòng)??期臨床試驗(yàn)���,2010年完成??期臨床試驗(yàn),2016 年完成???期臨床試驗(yàn)��。1997 年獲批開展臨床試驗(yàn)后���,對(duì)復(fù)方丹參滴丸質(zhì)量標(biāo)準(zhǔn)進(jìn)行深入研究��,例如增加含量測(cè)定�����、指紋圖譜�����、開展含量均勻度���、重金屬和農(nóng)殘檢查等,制定原藥材��、半成品、成品的質(zhì)量標(biāo)準(zhǔn)或質(zhì)量控制方法[6-14]��;生產(chǎn)全程符合GMP 規(guī)范��,藥材種植符合GAP規(guī)范要求[15-29]�����,2003 年丹參種植基地通過認(rèn)證[30-35]��。完成了FDA II 期臨床樣品溶出度�����、溶解度��、指標(biāo)成分含量測(cè)定方法及鑒別方法�����、溶劑殘留���、安全指標(biāo)成分等一系列檢驗(yàn)方法的研究工作[36-40]���,為藥效物質(zhì)基礎(chǔ)研究及指導(dǎo)工藝生產(chǎn)奠定基礎(chǔ)��,順利保證了??期臨床試驗(yàn)的成功[41-43]���。
II期臨床樣品和III 期臨床樣品均在符合GMP 條件下生產(chǎn)���。???期臨床試驗(yàn)前天士力全面實(shí)施了CMC 工藝再提升�����、質(zhì)量標(biāo)準(zhǔn)再完善[44-54]���,創(chuàng)新了制備工藝,創(chuàng)造了新技術(shù)和新裝備[55-56]�����,優(yōu)化了提取與制劑生產(chǎn)線�����、實(shí)施了廠房設(shè)備設(shè)施與工藝方法的全面再驗(yàn)證�����,提升了生產(chǎn)過程中的質(zhì)量控制手段[57-58]。CMC 確定了以提取物作為活性整體���,并參照化學(xué)原料藥方式設(shè)定提取物質(zhì)量標(biāo)準(zhǔn)��,通過實(shí)施提取物批次搭配(混合)投料提高指標(biāo)成分的控制精度���;USFDA 基于我國(guó)和EMA 相關(guān)指導(dǎo)原則要求,接受了含量均勻度的測(cè)定方法和穩(wěn)定性指標(biāo)的顯著性差異問題���;提取物標(biāo)準(zhǔn)除了強(qiáng)調(diào)常規(guī)的安全性檢查項(xiàng)目外��,還根據(jù)植物來源和藥效作用�����,增加側(cè)柏酮�����、硝酸鹽等檢測(cè)項(xiàng)目���;質(zhì)量一致性評(píng)價(jià)方面,藥材基源需鑒定至種及變種、亞種��,強(qiáng)調(diào)產(chǎn)地��、良好種植采收管理�����,藥材處理���、加工及儲(chǔ)存要遵循GMP 管理。根據(jù)工藝質(zhì)量要求��,強(qiáng)調(diào)批內(nèi)均勻性�����、質(zhì)量穩(wěn)定性��。對(duì)提取物及制劑��,要嚴(yán)格執(zhí)行USFDA 的cGMP 要求�����,在廠房、設(shè)備設(shè)施�����、工藝質(zhì)量設(shè)計(jì)及管理方面建立全面質(zhì)量管理與風(fēng)險(xiǎn)評(píng)估�����。含量控制指標(biāo)的建立與評(píng)價(jià)可結(jié)合藥理藥效���、臨床樣品設(shè)計(jì)及臨床試驗(yàn)的結(jié)果逐步提升與優(yōu)化�����。此外��,天士力集團(tuán)結(jié)合T89 的作用機(jī)理���、藥效特點(diǎn)探索開發(fā)了基于斑馬魚急性心肌缺血模型的生物效價(jià)方法;關(guān)于質(zhì)量平衡問題�����,USFDA 建議提供化學(xué)標(biāo)志物占提取物的重量百分比并對(duì)提取物中其他未明確的組分加以討論��,要求在上市前提取物和制劑的質(zhì)量標(biāo)準(zhǔn)中,設(shè)定大類組分的標(biāo)準(zhǔn)用于闡明質(zhì)量平衡���。USFDA 一方面認(rèn)可提取物活性整體的質(zhì)量控制復(fù)雜��,另一方面又希望通過化學(xué)組分的質(zhì)量平衡���、生物效價(jià)等研究來尋找到提取物活性整體的生物等效性評(píng)價(jià)方法,以方便產(chǎn)品生產(chǎn)過程質(zhì)量控制評(píng)價(jià)以及上市后的變更評(píng)價(jià)��。天士力在II 期臨床試驗(yàn)后�����,全面實(shí)施了符合USFDA 標(biāo)準(zhǔn)的cGMP 的質(zhì)量管理��。CMC 研究是基于質(zhì)量風(fēng)險(xiǎn)管理���,提倡質(zhì)量源于設(shè)計(jì)、依靠技術(shù)創(chuàng)新���、強(qiáng)化過程控制�����、實(shí)施全面驗(yàn)證���、持續(xù)完善標(biāo)準(zhǔn)的系統(tǒng)工程[59-60]���。
3.2 連花清瘟膠囊(KT07)
2013 年10 月以嶺藥業(yè)提交連花清瘟膠囊pre-IND 溝通交流,針對(duì)USFDA 正式回復(fù)��,制定工作方案��,包括藥材GAP 基地�����、藥品的CMC 到臨床���、藥理�����、藥學(xué)��、生產(chǎn)等環(huán)節(jié)�����。2015 年12 月獲批在美國(guó)開展II期臨床試驗(yàn)�����。
①以嶺制劑工藝優(yōu)化開展了以下橋接研究:陶瓷膜技術(shù)代替原醇沉除雜工藝(以膜通量�����、水提液出膏率以及有效成分和指紋圖譜為指標(biāo))��;真空帶式干燥技術(shù)代替噴霧干燥(對(duì)影響產(chǎn)品質(zhì)量的關(guān)鍵環(huán)節(jié)進(jìn)行考察)�����;濕法制粒變更為干法制粒(以顆粒得率�����、休止角和堆密度為指標(biāo));采用了動(dòng)態(tài)超聲逆流提取技術(shù)(以干膏收率��、有效成分含量���、指紋圖譜為考察指標(biāo))�����。
②藥材資源研究:美國(guó)要求藥材來源穩(wěn)定��,產(chǎn)地有明確的GPS 坐標(biāo)范圍�����,III期臨床試驗(yàn)和未來生產(chǎn)所用藥材均應(yīng)來自于所劃定的GPS 坐標(biāo)范圍內(nèi)��?����;氐倪x擇以氣候相似性為原則��,道地產(chǎn)區(qū)相對(duì)廣闊的一片區(qū)域�����,區(qū)域內(nèi)含3 個(gè)或3 個(gè)以上藥材基地�����,并遵循GACP 的規(guī)定��,提倡使用家種藥材,保證藥材的基原單一��。連花清瘟膠囊組方中的所有藥材均已固定產(chǎn)地區(qū)域和核心基地���,并采用傳統(tǒng)的植物學(xué)鑒定和現(xiàn)代的DNA 條形碼技術(shù)相結(jié)合保證基原準(zhǔn)確�����,在基地管理過程中遵循GACP���,由專人收集種植、管理���、采收加工�����,提供了基礎(chǔ)數(shù)據(jù)和影音資料���。
③CMC 研究:USFDA 建議在現(xiàn)有條件下,盡量采用多種分析和技術(shù)手段���,保證制劑間成分��、純度���、質(zhì)量、效力等一致��,避免藥物療效及安全性受影響���。USFDA 也強(qiáng)調(diào)嚴(yán)格的質(zhì)量控制應(yīng)從植物原材料到植物藥成品���,從非臨床研究到臨床試驗(yàn),貫穿始終���。從藥材源頭���、中間體到成品建立可溯源質(zhì)量控制體系。采用液相��、氣相��、紅外光譜等對(duì)藥材�����、中間體和成品進(jìn)行指紋圖譜和多成分含量測(cè)定研究。不要求列出所有成份���,只要求對(duì)植物原料藥質(zhì)量平衡��、有影響的大類成份進(jìn)行研究��,并在III期臨床前提供資料[61]�����。
4 美國(guó)已批準(zhǔn)植物藥的藥學(xué)研究情況
根據(jù)USFDA 公開的信息��,截至2014 年��,有超過600 個(gè)植物藥申報(bào)pre-INDs/INDs���,大約1/3 是商業(yè)申請(qǐng),2/3 是研究目的�����;其中2/3 是單個(gè)植物原材料�����,1/3 是多植物原材料,大部分只開展了II期臨床試驗(yàn)���,只有少數(shù)進(jìn)入了III期臨床試驗(yàn)。其中申報(bào)的適應(yīng)癥主要有腫瘤(34%)���;麻醉���、鎮(zhèn)痛和上癮(10%);皮膚病和牙科疾?����。?%)等[62]��。截至目前��,USFDA 批準(zhǔn)的新植物藥處方藥有4 個(gè)[63]��,見表2��。4 個(gè)產(chǎn)品中3 個(gè)是以NDA 途徑獲批�����,1 個(gè)是以BLA 途徑獲批。給藥途徑和劑型以外用或局部皮膚用凝膠或軟膏為主��,只有一個(gè)是口服緩釋片劑�����。除了Veregen 之外�����,其余3 個(gè)產(chǎn)品均獲得了多項(xiàng)新藥資格認(rèn)定�����。
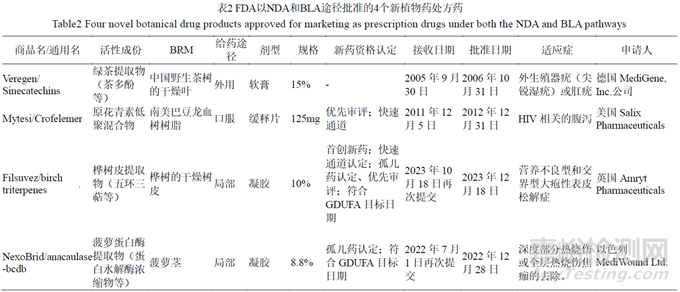
Filsuvez 和NexoBrid 在首輪審評(píng)時(shí)均未獲得批準(zhǔn)���。Filsuvez 首次申請(qǐng)是在2021 年3 月30 日��,2022 年2 月26 日收到完全回應(yīng)函(complete response letter��,CRL)�����,其中指出該申請(qǐng)未能提供支持Filsuvez 治療可以加速傷口愈合或減輕總傷口負(fù)擔(dān)的充分?jǐn)?shù)據(jù)���。為了解決這個(gè)問題�����,需要提交有關(guān)該產(chǎn)品治療EB(大皰性表皮松解癥)或EB 人群特定亞集的額外的確證性有效性證據(jù);Nexobrid 于2020 年6 月29 日首次提交申請(qǐng)��,由于產(chǎn)品質(zhì)量(product quality���,PQ)和科學(xué)調(diào)查辦公室(OSI)方面的諸多缺陷于2021 年6 月25 日收到CRL�����。從獲益-風(fēng)險(xiǎn)評(píng)估的角度��,關(guān)于PQ 缺陷�����,藥物質(zhì)量辦公室(office of product quality���,OPQ)的結(jié)論是�����,提交的數(shù)據(jù)不足以支持本品的生產(chǎn)是良好控制的��,生產(chǎn)的最終產(chǎn)品在貨架期內(nèi)是純凈的��、有效的��。OPQ 建議向申辦方簽發(fā)CRL 概述其缺陷并提出支持本品批準(zhǔn)所需要的信息和數(shù)據(jù)�����。其缺陷包括BRM 的真?zhèn)舞b定��、菠蘿蛋白酶的特殊生產(chǎn)工藝和原料藥微生物控制��、制劑微生物控制�����、制劑質(zhì)量方面的化學(xué)���、生產(chǎn)和控制等問題。此外��,批準(zhǔn)前需要對(duì)原料藥中間體生產(chǎn)車間、原料���、制劑以及凝膠溶媒生產(chǎn)車間進(jìn)行檢查�����,但由于旅行受限���,USFDA 無法在這輪審評(píng)中開展檢查。在本品再次提交后��,OPQ 和OSI 方面的問題得到解決�����,審評(píng)結(jié)論均建議批準(zhǔn)該申請(qǐng)���,見表2。本品于2023 年10 月16 日提交增加兒童人群的申請(qǐng)�����,2024 年8 月15 日獲批���。
根據(jù) USFDA 信息公開與保密的規(guī)定��,NDA 不得公開的信息包括生產(chǎn)方法或工藝��、質(zhì)量控制程序等[64]�����。本文基于4 個(gè)已批準(zhǔn)植物藥產(chǎn)品的藥學(xué)方面有限的公開信息進(jìn)行總結(jié)��,概括植物藥的藥學(xué)方面需要關(guān)注的主要問題���,舉例如下�����。①在BRM 方面�����,種屬的鑒別(鑒別種��、亞種和栽培品種等��,區(qū)分易混品��,提供真品證書)��、采收地理位置(eco-geographic regions���,EGRs)�����、加工過程的控制等���;GACP符合性,對(duì)采收人員的培訓(xùn)��,對(duì)BRM 建立質(zhì)量控制方法��,對(duì)農(nóng)藥殘留���、黃曲霉毒素、重金屬等雜質(zhì)進(jìn)行控制�����;采取措施��,加強(qiáng)環(huán)境的保護(hù);提供數(shù)據(jù)說明BRM 的天然變異程度�����。②應(yīng)建立原料藥和制劑穩(wěn)定性指示性的含量測(cè)定方法��,并規(guī)定上下限�����,可接受比化學(xué)藥更寬泛的含量限度��,但需要提供充分的依據(jù)��。③在質(zhì)量控制方面�����,應(yīng)建立雜質(zhì)檢查方法�����,并制定限度��;建立生物測(cè)定法確保批間一致性;建立溶出度和溶出曲線檢測(cè)方法�����。④對(duì)照品方面,需提供對(duì)照品的檢驗(yàn)報(bào)告�����、MSDS 等,對(duì)于自制對(duì)照品要提供確認(rèn)方案等�����。⑤含量測(cè)定��、鑒別等方法均應(yīng)經(jīng)過驗(yàn)證�����。⑥制定可重復(fù)和可靠的原料藥鑒別檢驗(yàn)方法�����、結(jié)構(gòu)鑒定和其他表征��,采用多種方法聯(lián)合測(cè)定表征��。⑦藥學(xué)研究與臨床有效性和安全性的相關(guān)性�����,采用與臨床終點(diǎn)相關(guān)的含量測(cè)定方法和指標(biāo)��。
5 討論與展望
根據(jù)對(duì) USFDA 植物藥相關(guān)指南�����、我國(guó)在USFDA 申報(bào)品種案例��、USFDA 已批準(zhǔn)產(chǎn)品的公開藥學(xué)研究?jī)?nèi)容的分析�����,總結(jié)美國(guó)植物藥注冊(cè)藥學(xué)研究的關(guān)鍵點(diǎn)和難點(diǎn),具體內(nèi)容如下:
5.1 申報(bào)過程中與USFDA 的溝通交流至關(guān)重要
各個(gè)研究階段申請(qǐng)人可就相關(guān)的問題與相關(guān)審評(píng)部門召開咨詢會(huì)或以其他途徑進(jìn)行咨詢���,例如:早期臨床試驗(yàn)植物原材料在種植和采收可能的變化和/或工藝優(yōu)化��,III期臨床試驗(yàn)處方的變化以及上市后種植基地���、種植和采收規(guī)范或加工方法等方面變更是否需要開展橋接研究,以及需要開展的橋接研究或其他研究類型��;早期臨床試驗(yàn)向制劑中加入單體活性成分的相關(guān)事宜�����;植物藥臨床試驗(yàn)中使用的安慰劑與試驗(yàn)藥物間有細(xì)微差別的相關(guān)問題��;多個(gè)適應(yīng)癥是否需要開發(fā)不同的生物測(cè)定方法等��。
5.2 分階段準(zhǔn)備藥學(xué)研究資料
對(duì)于有人用經(jīng)驗(yàn)的植物藥制劑��,在美國(guó)可直接開展II期或III期臨床試驗(yàn)���,II期臨床試驗(yàn)一般無需提交詳細(xì)的CMC 資料���,但在III期臨床試驗(yàn)啟動(dòng)前需要提交較完善的藥學(xué)研究資料。故在III期臨床試驗(yàn)前一般會(huì)開展工藝再提升��、質(zhì)量標(biāo)準(zhǔn)再完善��、生產(chǎn)車間的升級(jí)改造��、工藝驗(yàn)證等��,以及開發(fā)生物測(cè)定研究及質(zhì)量平衡法�����,以便變更的內(nèi)容或建立的質(zhì)控方法在III期臨床試驗(yàn)得到驗(yàn)證���。III期臨床試驗(yàn)后上市申請(qǐng)前不建議再有重大變更(例如原料藥的生產(chǎn)場(chǎng)地��、藥材和生產(chǎn)工藝等),以確保臨床試驗(yàn)用原料藥與擬上市用原料藥的一致性��,而無需開展橋接研究���。上市后對(duì)藥材產(chǎn)地���、種植和采收規(guī)范、和/或加工方法上所做的變更�����,均需要開展橋接研究。
5.3 生物測(cè)定研究
生物測(cè)定評(píng)價(jià)貼近臨床作用機(jī)制�����,通過將化學(xué)組成和活性建立關(guān)系���,從而評(píng)價(jià)原料藥的整體活性,它可作為生產(chǎn)過程質(zhì)量控制評(píng)價(jià)�����、穩(wěn)定性試驗(yàn)評(píng)價(jià)�����、不同階段原料藥相似性評(píng)價(jià)��、上市后變更評(píng)價(jià)及批間一致性評(píng)價(jià)的重要指標(biāo)���。作為一種質(zhì)控方法��,需要開展方法學(xué)驗(yàn)證和系統(tǒng)適用性研究以確保方法重復(fù)性和可預(yù)測(cè)性���。
5.4 確保制劑質(zhì)量一致性
質(zhì)量一致性評(píng)價(jià)包括對(duì)BRM 的控制�����、化學(xué)檢測(cè)的質(zhì)量控制和生產(chǎn)過程控制�����、生物測(cè)定和臨床數(shù)據(jù)的綜合評(píng)價(jià)�����,以證明上市樣品與上市前臨床試驗(yàn)用樣品的療效一致�����。BRM 控制包括實(shí)施DNA 條形碼鑒定技術(shù)�����、固定產(chǎn)地和基原���、符合GACP 等,GACP 無法覆蓋的種植�����、采收、加工和貯存等過程應(yīng)符合cGMP 要求���;提取物和制劑生產(chǎn)控制���,應(yīng)符合cGMP 要求,建立生物測(cè)定法整體控制質(zhì)量��,通過多批次�����、多劑量關(guān)鍵性臨床試驗(yàn)�����,確保研究藥物批次間臨床療效一致��。其中��,GACP���、cGMP 及臨床試驗(yàn)都需要大量的人力物力和時(shí)間投入,是注冊(cè)獲批的難點(diǎn)��。
5.5 植物藥作為藥品,具有常規(guī)藥品的一般要求��,同時(shí)也要遵循其自身特點(diǎn)
例如��,在藥品通性方面�����,資料提交要求與其他藥物相同���,需要以eCTD 格式遞交注冊(cè)申報(bào)資料��,植物原料可以以DMF 的形式或作為IND�����、NDA 資料的一部分遞交�����;采用穩(wěn)定性指示性的分析方法作為質(zhì)量控制方法�����;參照化學(xué)原料藥方式設(shè)定提取物質(zhì)量標(biāo)準(zhǔn)���,提取物批次混合投料提高指標(biāo)成分控制的精度��;原料和制劑要符合cGMP 等���。在遵循自身特點(diǎn)的方面,對(duì)植物來源起始物料的嚴(yán)格控制���;在穩(wěn)定性的顯著差異方面可以接受更寬泛的顯著性差異(>5%)�����;接受含量均勻度采用裝量或重量差異而非含量差異進(jìn)行測(cè)定;制定生物效價(jià)方法測(cè)定提取物(原料藥)整體活性���;采用多劑量多批次臨床試驗(yàn)考察不同藥材批次���、不同劑量的效應(yīng)關(guān)系等。
我國(guó)中藥藥學(xué)方面的技術(shù)指南經(jīng)過20 年的發(fā)展���,在總體原則方面已與美國(guó)相關(guān)要求一致[59]���,雖然可能還存在著理念�����、生產(chǎn)體系等方面的差異���,但相信在未來,若能選擇未被滿足的臨床需求�����、療效確切�����、處方簡(jiǎn)單�����、成份相對(duì)清楚���、國(guó)內(nèi)研究比較充分的中藥在美國(guó)開展植物藥處方藥注冊(cè)���,將大大提高注冊(cè)成功率��。
參考文獻(xiàn)
[1] FDA. What is botanical drug[EB/OL].(2025 -01-07)[2025-04-18]. https://www.fda.gov/about -fda/cder-offices-and-divisions/what-botanicaldrug.
[2] FDA .Frequently Asked Questions on Botanical Drug Product Development[EB/OL]. (2025 -01 questions-botanical-drug-productdevelopment.
[3] Freddie A H. Botanicals as “new” drugs: US development[J]. EPILEPSY Behav, 2015,52:338.
[4] FDA. Botanical drug development, guidance for industry[EB/OL].(2016 -12)[2025-02-01]. https://www.fda.gov/media/93113/download.
[5] 張萬良�����, 胡澤萍�����,周立紅�����,等.中國(guó)��、美國(guó)和歐盟植物藥注冊(cè)法規(guī)和技術(shù)要求

