近日����,FDA發(fā)布了PROGRAM 7356.002F《API 檢查指南》,文件為 API 生產(chǎn)工廠提供全面的 CGMP 檢查范圍�,并于2025年9月2日實(shí)施。
文件包含以下內(nèi)容:
PART I – BACKGROUND
第一部分
背景
1. Introduction
介紹
2. Applicable Statute, Regulations, and Guidances
適用法律、法規(guī)和指南
3. Scope of APIs Covered by This Program
本程序涵蓋的 API 范圍
4. Definitions
定義
PART II –IMPLEMENTATION
第二部分
實(shí)施
1. Objective
目的
2. Program Management Instructions
程序管理說(shuō)明
A. Selecting Sites
場(chǎng)地選擇
B. Selecting Investigators
檢查員的選擇
C. Selecting Inspection Types
檢查類(lèi)型的選擇
D. Selecting Systems
被檢查系統(tǒng)的選擇
E. Selecting APIs
API選擇
F. Selecting Profile Classes
配制文件類(lèi)型選擇
PART III –INSPECTIONAL
第三部分 檢查
1. Operations
操作
A. Inspection Approaches
檢查方法
B. System Coverage
被檢查系統(tǒng)的覆蓋范圍
C. Preparing the Inspection Strategy
準(zhǔn)備檢查策略
2. Reporting
報(bào)告
A. Special Instructions for Responding to a Form
FDA 483回復(fù) FDA 483 的特別說(shuō)明
PART IV –ANALYTICAL
第四部分 分析
PART V –REGULATORY/ADMINISTRATIVE STRATEGY
第五部分 監(jiān)管/行政策略
PART VI –REFERENCES, ATTACHMENTS, AND PROGRAM CONTACTS
第六部分 參考文獻(xiàn)�����、附錄和程序聯(lián)系方式
1. References
參考文獻(xiàn)
2. Attachments– None
附錄 無(wú)
3. Program Contacts
程序聯(lián)系方式
A. CDER
B. OII
文件詳細(xì)介紹了質(zhì)量體系、設(shè)施與設(shè)備系統(tǒng)����、生產(chǎn)系統(tǒng)、物料系統(tǒng)���、包裝與標(biāo)簽系統(tǒng)和實(shí)驗(yàn)室控制系統(tǒng)的檢查內(nèi)容:
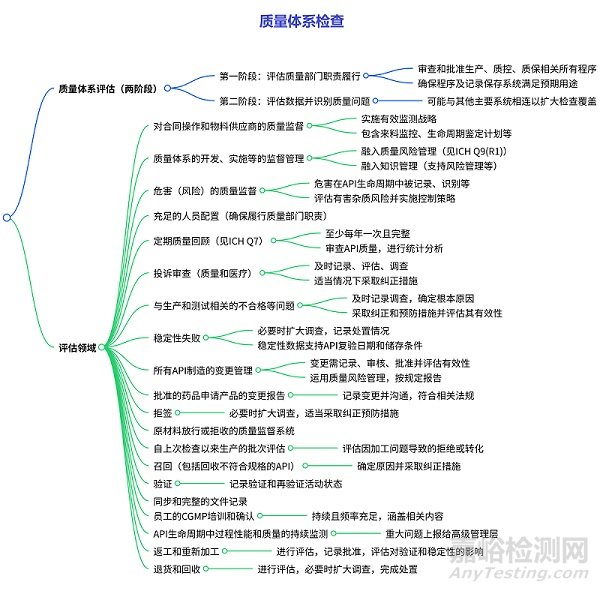
質(zhì)量系統(tǒng)的檢查
分為兩個(gè)階段:
第一階段:調(diào)查人員評(píng)估質(zhì)量部門(mén)是否履行了審查和批準(zhǔn)與生產(chǎn)�、質(zhì)量控制和質(zhì)量保證相關(guān)的所有程序的責(zé)任����,同時(shí)評(píng)估質(zhì)量部門(mén)是否確保這些程序及相關(guān)記錄保存系統(tǒng)足以滿(mǎn)足預(yù)期用途。
第二階段:調(diào)查人員對(duì)數(shù)據(jù)進(jìn)行評(píng)估�,識(shí)別任何質(zhì)量問(wèn)題,且該階段可能與其他主要系統(tǒng)相連以擴(kuò)大檢查覆蓋范圍��。
涵蓋多個(gè)方面��,包括對(duì)合同操作和物料供應(yīng)商的質(zhì)量監(jiān)督(需實(shí)施有效監(jiān)測(cè)戰(zhàn)略等)�;質(zhì)量體系開(kāi)發(fā)、實(shí)施等的監(jiān)督管理(融入質(zhì)量風(fēng)險(xiǎn)管理和知識(shí)管理)�����;危害(如交叉污染����、有害雜質(zhì))的質(zhì)量監(jiān)督(在 API 生命周期中進(jìn)行記錄�����、評(píng)估等)�����;充足的人員配置�����;定期質(zhì)量回顧(至少每年一次,含 API 質(zhì)量審查和統(tǒng)計(jì)分析)����;投訴審查(及時(shí)記錄、調(diào)查等)�;生產(chǎn)和測(cè)試相關(guān)的不合格等問(wèn)題的處理(及時(shí)調(diào)查、采取糾正預(yù)防措施)�;穩(wěn)定性失敗的應(yīng)對(duì)(擴(kuò)大調(diào)查、記錄處置等)��;API 制造的變更管理(記錄��、審核、評(píng)估風(fēng)險(xiǎn)等)��;批準(zhǔn)藥品的變更報(bào)告(符合法規(guī))���;拒簽��、原材料放行 / 拒收的質(zhì)量監(jiān)督���;自上次檢查以來(lái)生產(chǎn)批次的評(píng)估;召回(確定原因�、采取糾正措施);驗(yàn)證(記錄相關(guān)活動(dòng)狀態(tài))����;同步完整的文件記錄;員工 CGMP 培訓(xùn)(持續(xù)且涵蓋相關(guān)內(nèi)容)�����;API 生命周期中過(guò)程性能和質(zhì)量的監(jiān)測(cè)(重大問(wèn)題上報(bào))����;返工和重新加工(評(píng)估、記錄等);退貨和回收(評(píng)估��、調(diào)查等)��。同時(shí)���,文件多次引用 ICH 相關(guān)指南及 FDA 法規(guī)作為參考依據(jù)��。
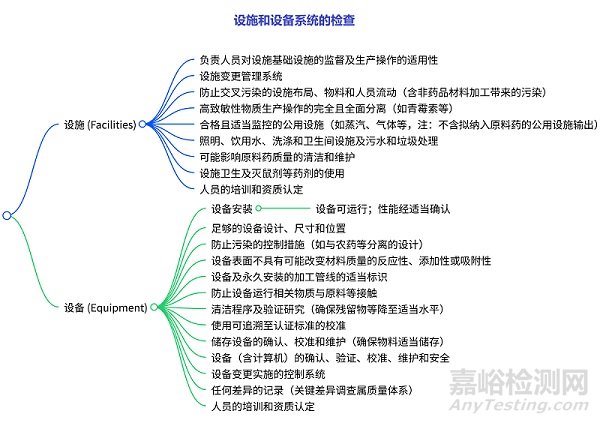
設(shè)施和設(shè)備系統(tǒng)的檢查
設(shè)施檢查:涉及人員監(jiān)督�、變更管理、布局與防污染����、高致敏性物質(zhì)生產(chǎn)隔離�����、公用設(shè)施監(jiān)控�、基礎(chǔ)設(shè)施(照明、水系統(tǒng)等)��、清潔維護(hù)���、衛(wèi)生與藥劑使用�����、人員培訓(xùn)等��。設(shè)備檢查:包括安裝與性能����、設(shè)計(jì)尺寸位置、防污染控制��、表面特性�、標(biāo)識(shí)、防物質(zhì)接觸����、清潔驗(yàn)證����、校準(zhǔn)標(biāo)準(zhǔn)、儲(chǔ)存設(shè)備維護(hù)��、設(shè)備全流程管理(含計(jì)算機(jī))、變更控制��、差異記錄�、人員培訓(xùn)等。
物料系統(tǒng)的檢查
涵蓋人員培訓(xùn)與資質(zhì)�����、物料及容器的標(biāo)識(shí)����、儲(chǔ)存、隔離與先進(jìn)先出使用�����,代表性樣品采集與檢驗(yàn)�����,關(guān)鍵物料供應(yīng)商審計(jì)監(jiān)控����,不合格物料處理,工藝用水和氣體系統(tǒng)適用性����,計(jì)算機(jī)化流程的確認(rèn)驗(yàn)證與安全,物料處理變更管理�,批次分發(fā)及差異記錄,以及起始物料等的風(fēng)險(xiǎn)管理計(jì)劃等內(nèi)容的檢查��。
生產(chǎn)系統(tǒng)的檢查
包含人員培訓(xùn)����、生產(chǎn)程序管理、關(guān)鍵活動(dòng)控制�����、偏差處理��、產(chǎn)量監(jiān)控��、工藝驗(yàn)證��、API 安全控制��、生產(chǎn)時(shí)限、設(shè)備標(biāo)識(shí)��、規(guī)格合理性�����、過(guò)程控制與檢測(cè)�、物料回收、交叉污染防控��、自動(dòng)化流程驗(yàn)證�、統(tǒng)計(jì)評(píng)估、變更管理����、批記錄管理及有害雜質(zhì)風(fēng)險(xiǎn)控制等的檢查,詳細(xì)列出了需要評(píng)估的內(nèi)容及相關(guān)要求����。
包裝和標(biāo)簽系統(tǒng)的檢查
包含人員培訓(xùn)資質(zhì)���、物料驗(yàn)收���、程序制定與執(zhí)行、變更管理�����、標(biāo)簽儲(chǔ)存與控制�����、記錄管理��、成品檢查����、有效期標(biāo)注、操作驗(yàn)證�、差異記錄及重新包裝要求等多個(gè)方面的檢查。
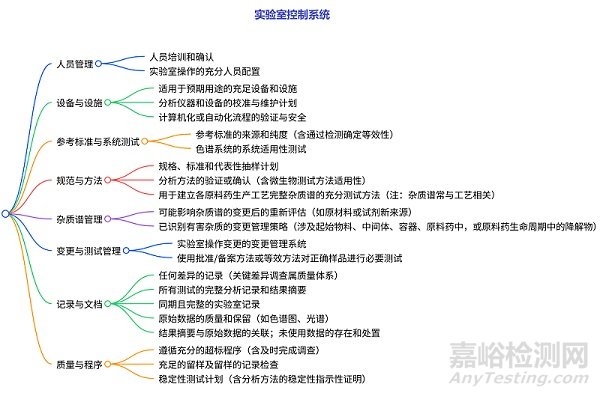
實(shí)驗(yàn)室控制系統(tǒng)的檢查
包含人員管理��、設(shè)備與設(shè)施(配置����、校準(zhǔn)維護(hù)、自動(dòng)化流程安全)��、參考標(biāo)準(zhǔn)與系統(tǒng)測(cè)試(來(lái)源純度、色譜系統(tǒng)適用性)�、規(guī)范與方法(抽樣計(jì)劃、方法驗(yàn)證�����、雜質(zhì)譜測(cè)試)�����、雜質(zhì)譜管理(變更后重評(píng)估�、有害雜質(zhì)變更策略)、變更與測(cè)試管理(操作變更系統(tǒng)���、樣品測(cè)試要求)����、記錄與文件(差異記錄�、分析記錄等)、質(zhì)量與程序(超標(biāo)處理��、留樣�、穩(wěn)定性測(cè)試)等多方面的檢查,明確了需要評(píng)估的內(nèi)容及相關(guān)要點(diǎn)��。
翻譯如下:
Active Pharmaceutical ingredient Process InspectionAPI
工藝檢查
PART I – BACKGROUND
第一部分 -背景
1. Introduction
引言
This compliance program applies a risk-based inspection strategy for surveillance inspections of API manufacturing facilities. The procedures in this compliance program maximize the use of history associated with a particular firm’s operations. These risks can include the firm’s compliance resources, the technology employed, the purported characteristics of the API, and the intended use of the API in the drug product, if known. This compliance program also provides procedures for coverage of for-cause inspections.
本合規(guī)計(jì)劃對(duì)活性藥物成分(API)生產(chǎn)設(shè)施的監(jiān)督檢查采用基于風(fēng)險(xiǎn)的檢查策略����。本合規(guī)計(jì)劃中的程序最大程度利用與某一特定公司運(yùn)營(yíng)相關(guān)的歷史情況���。這些風(fēng)險(xiǎn)可包括公司的合規(guī)資源��、所采用的技術(shù)�、API 宣稱(chēng)的特性�����,以及(若已知)API 在藥品中的預(yù)期用途��。本合規(guī)計(jì)劃還規(guī)定了有因檢查的覆蓋程序�。
An API is:
活性藥物成分(API)是:
[Any component that is intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease, or to affect the structure or any function of the body of man or other animals. The term includes those components that may undergo chemical change in the manufacture of the drug product and be present in the drug product in modified form intended to furnish the specified activity or effect.]
[旨在在疾病的診斷、治愈���、減輕�����、治療或預(yù)防中提供藥理活性或其他直接作用����,或影響人類(lèi)或其他動(dòng)物身體的結(jié)構(gòu)或任何功能的任何成分。該術(shù)語(yǔ)包括那些在藥品生產(chǎn)過(guò)程中可能發(fā)生化學(xué)變化�,并以旨在提供特定活性或作用的修飾形式存在于藥品中的成分。]
API production processes commonly include a series of operations. Major operations or steps in an API production process may include multiple chemical synthesis and fermentation, purification, crystallization, drying, milling, packing, labeling, and testing. This compliance program applies to all API manufacturing operations at the establishment, including receipt of materials, production, packaging, repackaging, labeling, relabeling, quality control, release, contract testing, storage and distribution of APIs, and related controls.
API 生產(chǎn)流程通常包括一系列操作���。API 生產(chǎn)流程中的主要操作或步驟可能包括多重化學(xué)合成與發(fā)酵�、純化�����、結(jié)晶�����、干燥�、研磨、包裝�、貼標(biāo)和測(cè)試。本合規(guī)計(jì)劃適用于機(jī)構(gòu)內(nèi)的所有 API 生產(chǎn)操作��,包括物料接收��、生產(chǎn)、包裝�、重新包裝、貼標(biāo)�����、重新貼標(biāo)����、質(zhì)量控制����、放行、合同測(cè)試��、API 的儲(chǔ)存和分銷(xiāo)����,以及相關(guān)控制。
2. Applicable Statute, Regulations, and Guidances
適用的法規(guī)��、規(guī)章和指南
Under section 510 of the FD&C Act,? API manufacturers must register their establishments with FDA and list their APIs in commercial distribution unless exempted under 21 CFR 207.13. Foreign drug manufacturers are also required to register their establishments and list all drugs imported or offered for import into the United States. Refer to 21 CFR 207.25 (h) for additional information on establishment registration requirements for foreign drug establishments.
根據(jù)《聯(lián)邦食品����、藥品和化妝品法案》(FD&C 法案)第 510 條,?API 制造商必須向 FDA 注冊(cè)其機(jī)構(gòu),并列出其商業(yè)分銷(xiāo)中的 API��,除非符合 21 CFR 207.13 的豁免規(guī)定�����。外國(guó)藥品制造商也必須注冊(cè)其機(jī)構(gòu)�����,并列出所有進(jìn)口到或擬進(jìn)口到美國(guó)的藥品�����。有關(guān)外國(guó)藥品機(jī)構(gòu)的注冊(cè)要求��,可參考 21 CFR 207.25 (h) 獲取更多信息��。
APIs are subject to the adulteration provisions of section 501 (a)(2)(B) of the FD&C Act, which requires that all drugs are manufactured in conformance with CGMP requirements. No distinction is made between an API and a drug product in the FD&C Act, and if either fails to comply with CGMP requirements for the product, FDA has no published compliance policy specifying a requirement for APIs or drug products. Thus, the CGMP requirements in CGMP regulations are frequently cited in the FD&C Act rather than the requirements for drug products in parts 210 and 211 (21 CFR parts 210 and 211).
API 受《聯(lián)邦食品����、藥品和化妝品法案》第 501 (a)(2)(B) 條有關(guān)摻假規(guī)定的約束,該條要求所有藥品的生產(chǎn)符合現(xiàn)行良好生產(chǎn)規(guī)范(CGMP)要求����。《聯(lián)邦食品、藥品和化妝品法案》中并未對(duì) API 和藥品制劑加以區(qū)分��,且若其中任何一種未符合產(chǎn)品的 CGMP 要求�,F(xiàn)DA 尚未發(fā)布明確針對(duì) API 或藥品制劑的合規(guī)政策。因此�����,《聯(lián)邦食品��、藥品和化妝品法案》中經(jīng)常引用 CGMP 法規(guī)中的 CGMP 要求�����,而非第 210 和 211 部分(21 CFR 第 210 和 211 部分)中針對(duì)藥品制劑的要求�。
FDA has long recognized that the concepts in the CGMP requirements for drug products in parts 210 and 211 are valid and applicable for API manufacturing. These concepts include: (1) ensuring drug quality by using suitable equipment and employing appropriately qualified and trained personnel; (2) establishing adequate written procedures and controls designed to ensure that manufacturing processes and controls are valid; (3) establishing a system of tests for in-process materials and drug products; and (4) ensuring that drug products are stable for their intended period of use.
FDA 長(zhǎng)期認(rèn)可 21 CFR 第 210 和 211 部分中成品藥的 CGMP 理念適用于 API 生產(chǎn)����,包括:(1)通過(guò)使用適宜設(shè)備和資質(zhì)合格的人員確保藥品質(zhì)量;(2)制定充分的書(shū)面程序和控制措施以保證生產(chǎn)過(guò)程及控制有效�;(3)建立中間物料和成品藥的檢測(cè)體系;(4)確保藥品在預(yù)期使用期限內(nèi)穩(wěn)定����。
FDA expects API manufacturers to: (1) apply CGMP to the API production process beginning with the use of starting materials; and (2) validate critical process steps that affect the quality and purity of the final API. The appropriate level of control is highly dependent on the manufacturing process and increases throughout the process as it proceeds from early steps to final isolation and purification steps. The appropriate level of control depends on the risk or criticality associated with each specific process step.FDA 要求 API 制造商:(1)從起始物料使用階段即對(duì) API 生產(chǎn)過(guò)程實(shí)施 CGMP;(2)驗(yàn)證影響最終 API質(zhì)量和純度的關(guān)鍵工藝步驟?���?刂扑饺Q于生產(chǎn)工藝,且隨流程從早期步驟推進(jìn)至最終分離純化步驟而逐步提高��,具體取決于各步驟的風(fēng)險(xiǎn)或關(guān)鍵程度��。
In 2001, FDA published the ICH guidance for industry Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients. ICH Q7 represents FDA’s current thinking on CGMP for APIs. Thus, API and related manufacturing and testing facilities that follow this guidance will generally be considered in compliance with the statutory CGMP requirement. However, alternative approaches may be used if such approaches satisfy the requirements of section 501(a)(2)(B) of the FD&C Act and ensure that the API meets its purported purity, identity, and quality characteristics.
FDA 于 2001 年發(fā)布 ICH 行業(yè)指南 Q7《活性藥物成分良好生產(chǎn)規(guī)范》��。ICH Q7 代表 FDA 對(duì) API 的CGMP 現(xiàn)行觀點(diǎn)����,因此遵循該指南的 API 生產(chǎn)及檢測(cè)設(shè)施通常被視為符合法定 CGMP 要求。但若替代方法能滿(mǎn)足《FD&C 法案》第 501 (a)(2)(B) 條并確保 API 符合其聲稱(chēng)的純度、標(biāo)識(shí)及質(zhì)量特性����,亦可采用����。
ICH Q7 provides guidance to industry on the extent and application of CGMP for manufacturing APIs under an appropriate quality system. The recommendations in ICH Q7 are also intended to help ensure that APIs meet the quality and purity characteristics that they purport or are represented to possess. Investigators should use ICH Q7 as a guideline for inspecting API manufacturers and related facilities.
ICH Q7 為行業(yè)提供在適宜質(zhì)量體系下 API 生產(chǎn)的 CGMP 范圍及應(yīng)用指南,其建議旨在確保 API 符合聲稱(chēng)的質(zhì)量和純度特性��。檢查人員應(yīng)將 ICH Q7 作為檢查 API 制造商及相關(guān)設(shè)施的指導(dǎo)文件����。
As part of FDA’s continued efforts to advance the Pharmaceutical Quality for the 21st Century initiative, FDA is pursuing strategies to ensure the implementation of an effective pharmaceutical quality system as described in the ICH guidance for industry Q10 Pharmaceutical Quality System (April 2009). In addition, FDA published the ICH guidance for industry Q9(R1) Quality Risk Management (May 2023) to provide principles and examples of tools for quality risk management that can be applied to different aspects throughout a product's lifecycle to ensure pharmaceutical quality. To facilitate the management of postapproval chemistry, manufacturing, and controls changes in a more predictable and efficient manner, FDA published the ICH guidance for industry Q12 Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management and its Annexes (May 2021) and the draft guidance for industry ICH Q12: Implementation Considerations for FDA-Regulated Products (May 2021). Knowledge management, change management, and quality risk management principles should be applied to API manufacturing operations holistically to help ensure API quality. These principles are described in the guidance for industry Quality Systems Approach to Pharmaceutical CGMP Regulations (September 2006) in addition to ICH Q9(R1), Q10, and Q12.
作為 “21 世紀(jì)藥品質(zhì)量” 倡議的持續(xù)努力�,F(xiàn)DA 致力于推動(dòng)實(shí)施 ICH Q10《藥品質(zhì)量體系》(2009 年4月)所述的有效藥品質(zhì)量體系�����。此外���,F(xiàn)DA 發(fā)布 ICH Q9 (R1)《質(zhì)量風(fēng)險(xiǎn)管理》(2023年5月)����,提供全產(chǎn)品生命周期內(nèi)可應(yīng)用于各環(huán)節(jié)的質(zhì)量風(fēng)險(xiǎn)管理原則及工具示例�;為促進(jìn)上市后化學(xué)、生產(chǎn)和控制變更的可預(yù)測(cè)性和高效管理���,發(fā)布 ICH Q12《藥品生命周期管理的技術(shù)和監(jiān)管考量》及其附件(2021年 5月)和草案《ICH Q12:FDA 監(jiān)管產(chǎn)品的實(shí)施考量》(2021年5月)�����。知識(shí)管理、變更管理和質(zhì)量風(fēng)險(xiǎn)管理原則應(yīng)全面應(yīng)用于 API 生產(chǎn)����,以確保質(zhì)量���。這些原則詳見(jiàn)《藥品 CGMP 的質(zhì)量體系方法》(2006年9月)及 ICH Q9 (R1)、Q10����、Q12。
To enhance FDA’s regulation of API manufacturing and quality, FDA may use additional information sources to inform its regulatory oversight. This may include the following: (1) other inspections conducted by FDA (e.g., preapproval and postapproval inspections); (2) existing inspection reports requested from trusted foreign regulatory partners through mutual recognition agreements and other confidentiality agreements; and (3) remote regulatory assessments,including (a) records or other information requested directly from facilities and other inspected entities under section 704(a)(4) of the FD&C Act and (b) remote interactive evaluations conducted where appropriate.
為加強(qiáng) API 生產(chǎn)和質(zhì)量監(jiān)管����,F(xiàn)DA 可利用其他信息來(lái)源輔助監(jiān)管,包括:(1)FDA 開(kāi)展的其他檢查(如批準(zhǔn)前和批準(zhǔn)后檢查)����;(2)通過(guò)互認(rèn)協(xié)議及其他保密協(xié)議從可信外國(guó)監(jiān)管機(jī)構(gòu)獲取的現(xiàn)有檢查報(bào)告;(3)遠(yuǎn)程監(jiān)管評(píng)估��,包括(a)根據(jù)《FD&C 法案》第 704 (a)(4) 條直接向設(shè)施及其他被檢查實(shí)體索取的記錄或信息����,(b)在適當(dāng)情況下進(jìn)行的遠(yuǎn)程互動(dòng)評(píng)估。
3. Scope of APIs Covered by This Program
本程序涵蓋的 API 范圍
This compliance program applies to the manufacture of APIs for use in human drug products, including:
本程序適用于人用藥品 API 的生產(chǎn)��,包括:
Small molecule APIs and critical intermediates manufactured by chemical synthesis
化學(xué)合成的小分子 API 及關(guān)鍵中間體
Polypeptides consisting of up to 40 amino acids that are manufactured by chemical synthesis or fermentation
化學(xué)合成或發(fā)酵生產(chǎn)的含至多 40 個(gè)氨基酸的多肽
Antibiotics and other small molecule APIs produced by microbial fermentation
微生物發(fā)酵生產(chǎn)的抗生素及其他小分子API
This compliance program does not cover APIs for blood, vaccines, allergenics, tissues, and cellular and gene therapies. These products are regulated by the Center for Biologics Evaluation and Research. CGMP inspections of APIs that are proteins are conducted using compliance program 7356.002M-Surveillance Inspections of Protein Drug Substance Manufacturers. Examples of biological products that contain APIs that are proteins include, but are not limited to:
本程序不涵蓋血液、疫苗����、變應(yīng)原�����、組織及細(xì)胞和基因治療用 API���,此類(lèi)產(chǎn)品由生物制品評(píng)價(jià)與研究中心監(jiān)管�����。蛋白質(zhì)類(lèi) API 的 CGMP 檢查遵循合規(guī)程序 7356.002M 《蛋白質(zhì)活性藥物成分制造商的監(jiān)督檢查》。含蛋白質(zhì)類(lèi) API 的生物制品示例包括但不限于:
Enzymes
酶
Monoclonal antibodies
單克隆抗體
Antibody-drug conjugates
抗體藥物偶聯(lián)物
Fusion proteins (e.g., antibody Fc region-containing fusion proteins)
融合蛋白(如含抗體 Fc 區(qū)的融合蛋白)
Growth factors
生長(zhǎng)因子
Cytokines (e.g., interleukins, interferons, tumor necrosis factors)
細(xì)胞因子(如白細(xì)胞介素、干擾素�����、腫瘤壞死因子)
Botulinum toxins
肉毒毒素
Insulin and insulin analogues
胰島素及其類(lèi)似物
Synthetically derived proteins
合成衍生蛋白質(zhì)
This compliance program applies to the manufacture of APIs intended to be sterile only up to the point immediately before the API is rendered sterile. However, neither this compliance program nor ICH Q7 provide guidance on the sterilization and aseptic processing for sterile APIs. Investigators should use compliance program 7356.002A-Sterile Drug Process Inspections and the guidance for industry on aseptic processing, Sterile Drug Products Produced by Aseptic Processing-Current Good Manufacturing Practice (September 2004), when inspecting the sterile processing of APIs that are purported to be sterile (including for compounding sterile preparations).
本程序適用于擬無(wú)菌 API 的生產(chǎn)��,但僅覆蓋至 API 滅菌前的環(huán)節(jié)��。本程序及 ICH Q7 均未提供無(wú)菌 API的滅菌及無(wú)菌工藝指南。檢查聲稱(chēng)無(wú)菌的 API 的無(wú)菌工藝(包括無(wú)菌配制制劑)時(shí)�����,檢查人員應(yīng)遵循合規(guī)程序 7356.002A《無(wú)菌藥品生產(chǎn)過(guò)程檢查》及《無(wú)菌工藝生產(chǎn)的無(wú)菌藥品 - 當(dāng)前良好生產(chǎn)規(guī)范》(2004 年 9月)�����。
Bulk finished products are manufactured similarly to APIs, but they do not undergo further processing or compounding after their synthesis, fermentation, or extraction and are instead repackaged into market containers. Bulk finished products are subject to the requirements of parts 210 and 211. The synthesis and fermentation processes that result in such APIs are covered by this program rather than the program for dosage forms (i.e., compliance program 7356.002-Drug Manufacturing Inspections).
散裝成品的生產(chǎn)方式與 API 類(lèi)似�����,但其合成���、發(fā)酵或提取后不再經(jīng)進(jìn)一步加工或配制,僅重新包裝至市售容器��。散裝成品需遵守 21 CFR 第 210 和 211 部分的要求��,其合成和發(fā)酵過(guò)程由本程序覆蓋�����,而非劑型相關(guān)程序(即合規(guī)程序 7356.002《藥品生產(chǎn)檢查》)。
PART II – IMPLEMENTATION
第二部分-實(shí)施
1. Objective
1目標(biāo)
The primary objective of this compliance program is to provide comprehensive CGMP inspectional coverage of API manufacturing establishments. This is done through system-based inspections that represent all profile classes (i.e., types of API manufacturing processes) and determine whether a manufacturer is operating in a state of control. An API manufacturer is operating in a state of control when it employs conditions and practices that ensure compliance with section 501(a)(2)(B) of the FD&C Act. A firm that is in a state of control produces APIs that have an adequate level of assurance of quality, identity, and purity.
本程序的主要目標(biāo)是全面覆蓋 API 生產(chǎn)機(jī)構(gòu)的 CGMP 檢查�����,通過(guò)基于系統(tǒng)的檢查涵蓋所有工藝類(lèi)別(即API 生產(chǎn)工藝類(lèi)型),并判定制造商是否處于受控狀態(tài)��。當(dāng) API 制造商的條件和實(shí)踐符合《FD&C 法案》第 501 (a)(2)(B) 條時(shí)���,即處于受控狀態(tài)�,其所生產(chǎn)的 API 具有充分的質(zhì)量、標(biāo)識(shí)和純度保證��。
A firm is not in a sufficient state of control if any system is significantly noncompliant with CGMP requirements such that the quality, identity, and purity of the API cannot be adequately ensured. Documented CGMP deficiencies provide the evidence for concluding that a system is not operating in a state of control. See Part V - Regulatory/Administrative Strategy, for a discussion of compliance actions based on inspection findings that demonstrate that a system or multiple systems are not in a state of control.
若任何系統(tǒng)嚴(yán)重違反 CGMP 要求����,導(dǎo)致無(wú)法充分保證 API 的質(zhì)量����、標(biāo)識(shí)和純度,則企業(yè)未處于充分受控狀態(tài)���。有記錄的 CGMP 缺陷是判定系統(tǒng)失控的依據(jù)?��;跈z查發(fā)現(xiàn)的單系統(tǒng)或多系統(tǒng)失控情況所采取的合規(guī)措施��,詳見(jiàn)第五部分《監(jiān)管 / 行政策略》�。
Profile classes generalize inspectional coverage from a small number of specific APIs to all APIs in that class. This compliance program uses a systems-based approach to further generalize inspectional coverage from a small number of profile classes to an overall evaluation of the firm. This allows preapproval inspections to focus on the specific issues related to a given drug application and improves the assessment process by providing timely and efficient support for drug application decisions.
工藝類(lèi)別將檢查覆蓋范圍從少量特定 API 擴(kuò)展至該類(lèi)別下所有 API。本程序采用基于系統(tǒng)的方法�����,進(jìn)一步將覆蓋范圍從少量工藝類(lèi)別擴(kuò)展至企業(yè)整體評(píng)估,使批準(zhǔn)前檢查可聚焦于特定藥品申請(qǐng)的相關(guān)問(wèn)題���,并為申請(qǐng)決策提供及時(shí)高效的支持。
Investigators should use this compliance program’s system definitions and organization when inspecting API manufacturers and reporting the results. Focusing on systems, rather than just profile classes, increases the efficiency of inspections, because the systems are oftenapplicable to multiple profile classes. An inspection under this program is profileable and will result in a determination of acceptability or nonacceptability for all API profile classes specified in this compliance program. Inspection coverage should represent all of the API profile classes that are manufactured by the firm.
檢查人員檢查 API 制造商及報(bào)告結(jié)果時(shí),應(yīng)采用本程序的系統(tǒng)定義和架構(gòu)�����。聚焦系統(tǒng)而非僅關(guān)注工藝類(lèi)別可提高檢查效率�,因系統(tǒng)通常適用于多個(gè)工藝類(lèi)別。本程序下的檢查可形成概況結(jié)論�,對(duì)所有指定的 API 工藝類(lèi)別做出可接受或不可接受的判定�,且覆蓋范圍應(yīng)代表企業(yè)生產(chǎn)的所有 API 工藝類(lèi)別。
The other objectives of this compliance program include:
本程序的其他目標(biāo)包括:
Determining whether inspected establishments are operating in compliance with applicable CGMP requirements to aid in FDA’s enforcement of the FD&C Act.
判定被檢查機(jī)構(gòu)是否遵守適用的 CGMP 要求����,以支持 FDA 執(zhí)行《FD&C 法案》��。
Initiating appropriate action against manufacturers that are found to be out of compliance.
對(duì)不合規(guī)制造商采取適當(dāng)措施�����。
Obtaining information that may affect other drug manufacturing or compounding operations(e.g., sterilization, drug product manufacturing, drug product compounding).
獲取可能影響其他藥品生產(chǎn)或配制操作(如滅菌����、成品藥生產(chǎn)����、藥品配制)的信息�。
Providing guidance to manufacturers during inspections to improve compliance with CGMPrequirements, as applicable.
檢查中為制造商提供指導(dǎo)�����,以提高其 CGMP 合規(guī)性(如適用)�。
2. Program Management Instructions
程序管理說(shuō)明
CDER uses a risk-based site selection model to identify API firms for OII’s routine surveillance inspections. CDER and OII maintain drug profiles per Chapter 5 of the Investigations Operations Manual (IOM).
CDER 采用基于風(fēng)險(xiǎn)的場(chǎng)地選擇模型�,為 OII 的常規(guī)監(jiān)督檢查確定 API 企業(yè)�����。CDER 和 OII 根據(jù)《調(diào)查操作手冊(cè)》(IOM)第 5 章維護(hù)藥品概況��。
Unless specifically directed by CDER, OII is responsible for determining the depth of inspectional coverage for each API firm using this compliance program’s instructions. CGMP inspectional coverage under this program will be sufficient to assess the state of compliance for each firm.
除非 CDER 另有明確指示,OII 負(fù)責(zé)根據(jù)本程序要求確定各 API 企業(yè)的檢查深度����。本程序下的 CGMP 檢查覆蓋范圍應(yīng)足以評(píng)估各企業(yè)的合規(guī)狀態(tài)��。
Inspections of API manufacturers should be conducted by experienced investigators with sufficient education and training related to the type of API production process or processes conducted by the API manufacturer (e.g., fermentation, chemical synthesis). Chemists and microbiologists should be considered for inclusion on API inspection teams, as appropriate, particularly for evaluating laboratory operations (e.g., analytical methods evaluation, analytical data, laboratory procedures, instrumentation) and assessing analytical methods that are used to establish impurity profiles, fermentation manufacturing processes, and complex multistep processes for chemical synthesis.
API 制造商的檢查應(yīng)由經(jīng)驗(yàn)豐富的檢查人員執(zhí)行,其需接受過(guò)與 API 生產(chǎn)工藝類(lèi)型(如發(fā)酵����、化學(xué)合成)相關(guān)的充分教育和培訓(xùn)。應(yīng)酌情納入化學(xué)家及微生物學(xué)家組成檢查團(tuán)隊(duì)���,尤其在評(píng)估實(shí)驗(yàn)室操作(如分析方法評(píng)價(jià)���、分析數(shù)據(jù)�����、實(shí)驗(yàn)室程序����、儀器)及用于建立雜質(zhì)概況���、發(fā)酵工藝和復(fù)雜多步化學(xué)合成的分析方法時(shí)。
Investigators conducting API inspections must understand the basic differences between theprocesses used to produce APIs and the processes used to produce finished dosage forms.APIs are usually produced by chemical synthesis or by cell culture and extraction. Thus, API production typically involves significant changes to the starting materials or intermediates by various chemical, physical, and biological processing steps. Generally, the ultimate objective in API production is to achieve a pure compound of certain identity. In contrast,the ultimate objective of finished dosage form manufacturing is generally to achieve the uniform distribution of an API across dosing units to deliver a precise amount of API.
執(zhí)行 API 檢查的檢查人員必須理解 API 與成品劑型生產(chǎn)工藝的基本差異。API 通常通過(guò)化學(xué)合成或細(xì)胞培養(yǎng)及提取生產(chǎn)��,其過(guò)程涉及通過(guò)化學(xué)�����、物理和生物步驟對(duì)起始物料或中間體進(jìn)行顯著改造��,最終目標(biāo)是獲得特定標(biāo)識(shí)的純化合物���。而成品劑型生產(chǎn)的最終目標(biāo)通常是實(shí)現(xiàn) API 在劑量單位中的均勻分布�����,以遞送精確劑量��。
There are two basic types of CGMP inspections: surveillance and for-cause. Surveillance inspections are conducted on a routine basis to satisfy FDA’s responsibilities to inspect drug manufacturing facilities. For-cause inspections are conducted in response to violative surveillance inspections and when a need arises to inspect a facility in response to specific events or information.
CGMP 檢查主要分為兩類(lèi):監(jiān)督檢查和針對(duì)性檢查。監(jiān)督檢查為常規(guī)檢查���,以履行 FDA 對(duì)藥品生產(chǎn)設(shè)施的檢查職責(zé);針對(duì)性檢查針對(duì)違規(guī)的監(jiān)督檢查結(jié)果,或在需響應(yīng)特定事件或信息時(shí)開(kāi)展��。
For-cause inspections include: (1) follow-up CGMP compliance inspections to verify corrective actions after a regulatory action has been taken; and (2) CGMP inspections in response to specific events or information (e.g., field alert reports, biological product defect reports, industry complaints, recalls, other indicators of defective products) that bring the compliance or quality of a manufacturing practice, facility, process, or API into question.
針對(duì)性檢查包括:(1)監(jiān)管措施后的后續(xù) CGMP 合規(guī)檢查,以驗(yàn)證糾正措施�����;(2)響應(yīng)特定事件或信息(如現(xiàn)場(chǎng)警報(bào)報(bào)告、生物制品缺陷報(bào)告����、行業(yè)投訴����、召回�、其他產(chǎn)品缺陷跡象)的 CGMP 檢查,此類(lèi)事件或信息使生產(chǎn)實(shí)踐�、設(shè)施、工藝或 API 的合規(guī)性或質(zhì)量受到質(zhì)疑。
Follow-up CGMP compliance inspections provide focused coverage, including the areas of concern, the proposed corrective action plan for affected operations, any implemented corrective actions, and/or the deficiencies noted on Form FDA 483–Inspectional Observations for a previous inspection. System coverage can be added on a case-by-case basis. Follow-up CGMP compliance inspections to a warning letter or other significant regulatory actions are also considered for-cause inspections and, as a result, the related for-cause assignmentscan request either full systems coverage or individual system coverage. In addition, coverage can be added on a case-by-case basis, at OII’s discretion, before or during the inspection.
后續(xù) CGMP 合規(guī)檢查聚焦于關(guān)注領(lǐng)域����、受影響操作的擬議糾正行動(dòng)計(jì)劃��、已實(shí)施的糾正措施及 / 或上一次檢查中 FDA 483 表《檢查觀察結(jié)果》所列缺陷,可酌情增加系統(tǒng)覆蓋。針對(duì)警告信或其他重大監(jiān)管措施的后續(xù)檢查亦屬針對(duì)性檢查,相關(guān)任務(wù)可要求全面系統(tǒng)覆蓋或單個(gè)系統(tǒng)覆蓋���,OII 可在檢查前或檢查中酌情增加覆蓋范圍。
Other for-cause inspections (e.g., inspections due to industry complaints or other indicators of defective APIs) may be initiated, but these inspections can be expanded to include CGMP coverage for the purpose of updating the firm’s overall compliance status.
其他針對(duì)性檢查(如因行業(yè)投訴或 API 缺陷跡象開(kāi)展的檢查)可擴(kuò)展至 CGMP 覆蓋范圍�,以更新企業(yè)整體合規(guī)狀態(tài)。
This is the general scheme of systems for inspecting API manufacturers:
以下是 API 制造商檢查的系統(tǒng)總體框架:
1.Quality System ensures overall compliance with CGMP requirements and internal procedures and specifications. A robust quality system relies on documentation and strong senior management oversight of CGMP operations and quality-related matters, supports and facilitates the activities conducted under all of the six systems, monitors its effectiveness, and ensures a commitment to an established quality policy.
質(zhì)量系統(tǒng):確保全面遵守 CGMP 要求及內(nèi)部程序和規(guī)格����。健全的質(zhì)量系統(tǒng)依賴(lài)于 CGMP 操作及質(zhì)量相關(guān)事項(xiàng)的文件記錄和強(qiáng)有力的高級(jí)管理層監(jiān)督��,支持并促進(jìn)其他六個(gè)系統(tǒng)的活動(dòng)���,監(jiān)控其有效性,并確保對(duì)既定質(zhì)量方針的承諾。
2.Facilities and Equipment System includes activities which provide an appropriate physical environment and equipment used in the production of APIs.
設(shè)施與設(shè)備系統(tǒng):包括提供適宜的物理環(huán)境及 API 生產(chǎn)所用設(shè)備的相關(guān)活動(dòng)。
3. Materials System includes measures and activities to control starting materials, intermediates, and containers.It includes validation of computerized and inventory control processes,storage, and distribution controls.
物料系統(tǒng):包括控制起始物料�����、中間體和容器的措施及活動(dòng)���,涵蓋計(jì)算機(jī)化和庫(kù)存控制系統(tǒng)��、儲(chǔ)存及分銷(xiāo)控制的驗(yàn)證。
4.Production System includes measures and activities to control the manufacture of APIs, including in-process sampling, testing, and process validation.
生產(chǎn)系統(tǒng):包括控制 API 生產(chǎn)的措施及活動(dòng),含過(guò)程中采樣��、測(cè)試和工藝驗(yàn)證�。
5.Packaging and Labeling System includes measures and activities that control the packaging and labeling of intermediates and APIs.包裝與貼標(biāo)簽系統(tǒng):包括控制中間體和 API 包裝及貼標(biāo)簽的措施及活動(dòng)。
6.Laboratory Control System includes measures and activities related to laboratory procedures, testing, analytical methods development, analytical methods validation or verification, and the stability program.
實(shí)驗(yàn)室控制系統(tǒng):包括與實(shí)驗(yàn)室程序�、測(cè)試、分析方法開(kāi)發(fā)�、分析方法驗(yàn)證或確認(rèn)及穩(wěn)定性方案相關(guān)措施及活動(dòng)。
Inspectional Section III.1.B has detailed inspection coverage guidance for these systems.
第三部分第 1.B 節(jié)提供這些系統(tǒng)的詳細(xì)檢查覆蓋指南�����。
An inspection under this program is defined as audit coverage of two or more systems, including mandatory coverage of the quality system. Inspecting at least two systems (i.e., the quality system and one other system), or more systems if deemed necessary by OII, will provide the basis for an overall inspection classification decision.
本程序下的檢查定義為對(duì)兩個(gè)或多個(gè)系統(tǒng)的審計(jì)覆蓋���,包括對(duì)質(zhì)量系統(tǒng)的強(qiáng)制性覆蓋。檢查至少兩個(gè)系統(tǒng)(即質(zhì)量系統(tǒng)及另一個(gè)系統(tǒng))�,或 OII 認(rèn)為必要時(shí)檢查更多系統(tǒng)��,將作為整體檢查分類(lèi)決策的依據(jù)。
Coverage of a system should be sufficiently detailed and specific examples should be selected. Sufficient coverage ensures that the system inspection outcome reflects the system’s state of control for every profile class. If a particular representative system is adequate, it should be adequate for all drug profile classes manufactured by the firm. In some circumstances, it may not be possible to generalize certain deficiencies in a system to all API profile classes. If so, the unaffected profile classes may be considered acceptable if they are otherwise acceptable.
對(duì)一個(gè)系統(tǒng)的覆蓋應(yīng)足夠詳細(xì)���,并選擇具體示例。充分覆蓋可確保系統(tǒng)檢查結(jié)果反映每個(gè)工藝類(lèi)別的系統(tǒng)受控狀態(tài)。若某個(gè)代表性系統(tǒng)是充分的,則其應(yīng)適用于企業(yè)生產(chǎn)的所有藥品工藝類(lèi)別�。在某些情況下,可能無(wú)法將系統(tǒng)中的特定缺陷推廣至所有 API 工藝類(lèi)別��,此時(shí)若未受影響的工藝類(lèi)別在其他方面符合要求����,則可視為可接受����。
If a selected API has a unique processing or control function that is part of a system that was not chosen for coverage, the unique function can be covered for that API. However, the system for the unique function does not need to be given full coverage. For example, if an API chosen for coverage uses only high purity water in its manufacture, the water purification system can be inspected without giving full inspection coverage to the materials system. Selecting unique functions within a system will be at the discretion of the investigator.
若所選 API 具有屬于未被選擇覆蓋的系統(tǒng)的獨(dú)特加工或控制功能����,可針對(duì)該 API 覆蓋此獨(dú)特功能��,但無(wú)需全面覆蓋該功能所屬的系統(tǒng)��。例如�����,若所選 API 生產(chǎn)僅使用高純水�����,可檢查水純化系統(tǒng)�,而無(wú)需全面覆蓋物料系統(tǒng)。檢查人員可自行決定是否選擇系統(tǒng)內(nèi)的獨(dú)特功能���。
Complete inspection of one system may require follow up of certain aspects of another system to fully document the findings. However, this coverage does not constitute nor require complete coverage of the other system.
完整檢查一個(gè)系統(tǒng)可能需要跟進(jìn)另一個(gè)系統(tǒng)的某些方面以充分記錄發(fā)現(xiàn)���,但此類(lèi)覆蓋不構(gòu)成也不要求對(duì)另一個(gè)系統(tǒng)進(jìn)行完整覆蓋。
Inspections should cover any APIs referenced in the assignment and, as appropriate, any other representative APIs based on their level of risk. For foreign API firms, investigators should cover only APIs that are marketed or intended to be marketed in the United States.
檢查應(yīng)覆蓋任務(wù)中提及的所有 API����,并酌情覆蓋基于風(fēng)險(xiǎn)水平的其他代表性 API。對(duì)于外國(guó) API 企業(yè)�����,檢查人員僅需覆蓋在美上市或擬在美上市的 API��。
Investigators should select APIs so that the coverage represents the establishment’s overall ability to manufacture in compliance with CGMP requirements. API selection should also be based on the level of risk, including selection of APIs that are:
檢查人員選擇的 API 應(yīng)能體現(xiàn)機(jī)構(gòu)遵守 CGMP 要求的整體生產(chǎn)能力����,且選擇應(yīng)基于風(fēng)險(xiǎn)水平�,包括以下API:
Used in approved drug products
用于已批準(zhǔn)藥品的 API
Therapeutically significant
具有治療重要性的 API
Difficult to manufacture
難以生產(chǎn)的 API
Intended for use in parenteral or modified-release products or in combination products
擬用于注射劑、調(diào)釋制劑或組合產(chǎn)品的 API
Documented as having past compliance problems
有記錄顯示存在合規(guī)問(wèn)題的 API
However, this does not prevent investigators from selecting less therapeutically significant APIs to evaluate specific APIs (or profile classes) that were not previously given in-depthcoverage at the facility. Inspection coverage depth and intensity can be reduced for these APIs unless deficiencies are identified.
但這并不妨礙檢查人員選擇治療重要性較低的 API�,以評(píng)估設(shè)施中先前未被深入覆蓋的特定 API(或工藝類(lèi)別)�。除非發(fā)現(xiàn)缺陷�����,否則這些 API 的檢查深度和強(qiáng)度可降低��。
When a system is inspected, the inspection of that system generally applies to all API products that use the system. Because API manufacturers are often referenced in multiple drug applications, each inspection should cover an adequate number and type of APIs to cover the selected systems. For example, if an inspection covers the production system for a site making one API by fermentation and another by synthesis, the inspection should include both types of processing in the physical inspection and the records that are sampled during the audit. This strategy, together with the classification of all applicable profile classes after the inspection, will maximize the use of FDA resources and avoid repeated visits to the same manufacturing site to cover different API profile classes.
檢查一個(gè)系統(tǒng)時(shí)�����,該系統(tǒng)的檢查結(jié)果通常適用于所有使用該系統(tǒng)的 API 產(chǎn)品����。由于 API 制造商常被多個(gè)藥品申請(qǐng)引用,每次檢查應(yīng)覆蓋足夠數(shù)量和類(lèi)型的 API 以覆蓋所選系統(tǒng)����。例如,若檢查覆蓋某場(chǎng)地通過(guò)發(fā)酵生產(chǎn)一種 API 和通過(guò)合成生產(chǎn)另一種 API 的生產(chǎn)系統(tǒng)����,則實(shí)地檢查和審計(jì)抽樣記錄應(yīng)包含這兩種工藝類(lèi)型。該策略結(jié)合檢查后對(duì)所有適用工藝類(lèi)別的分類(lèi)�,可最大限度利用 FDA 資源,避免為覆蓋不同 API 工藝類(lèi)別而多次訪問(wèn)同一生產(chǎn)場(chǎng)地�����。
Profile class codes or APIs selected for coverage should represent all of the APIs manufactured at the firm. Profile class codes may also be grouped by similarity, such that coverage of one profile class is sufficient to demonstrate the CGMP conditions for another profile class. For example, inspecting a profile class code of CSS could provide surrogate coverage of CSN. Similarly, inspecting a profile class code of CBI could provide surrogate coverage of other profile classes, such as CFN, CFS, and perhaps CEX.
所選工藝類(lèi)別代碼或 API 應(yīng)代表企業(yè)生產(chǎn)的所有 API。工藝類(lèi)別代碼可按相似性分組��,因此對(duì)一個(gè)工藝類(lèi)別的覆蓋足以證明另一個(gè)工藝類(lèi)別的 CGMP 狀況�。例如,檢查 CSS 工藝類(lèi)別代碼可替代覆蓋 CSN�;同樣,檢查 CBI 工藝類(lèi)別代碼可替代覆蓋 CFN����、CFS 或許還有 CEX 等其他工藝類(lèi)別。
The inspection findings will be used to update all applicable profile classes. Normally, an inspection under this system approach will result in all applicable profile classes being updated. For more information, see Exhibit 5-14 Profiling a Firm’s CGMP/QS Compliance Status in the IOM.
檢查結(jié)果將用于更新所有適用的工藝類(lèi)別�����。通常�����,基于此系統(tǒng)方法的檢查將更新所有適用的工藝類(lèi)別��。更多信息參見(jiàn)《調(diào)查操作手冊(cè)》中的附件 5-14《企業(yè) CGMP/QS 合規(guī)狀態(tài)概況》���。
PART III – INSPECTIONAL
第三部分-檢查
1. Operations
操作
Investigators conducting API inspections should understand the general inspection strategy in this compliance program. API firms vary greatly in size, diversity of operations, and quality assurance systems, so investigators should carefully plan their inspection strategy at each firm. See Part III.1.C for the procedures for preparing an inspection strategy.
進(jìn)行 API 檢查的檢查人員應(yīng)理解本合規(guī)程序中的總體檢查策略��。API 企業(yè)在規(guī)模���、運(yùn)營(yíng)多樣性和質(zhì)量保證體系方面差異很大,因此檢查人員應(yīng)仔細(xì)規(guī)劃在每個(gè)企業(yè)的檢查策略���。制定檢查策略的程序詳見(jiàn)第三部分第 1.C節(jié)��。
Investigators should also review the firm’s rationale for the point at which API production begins, as described in ICH Q7 and the ICH guidance for industry Q11 Development and Manufacture of Drug Substances (November 2012). This point may vary based on the type of process used for the API (e.g., synthesis, fermentation, extraction, purification).
檢查人員還應(yīng)審核企業(yè)關(guān)于 API 生產(chǎn)起始點(diǎn)的理由��,如 ICH Q7 及 ICH 行業(yè)指南 Q11《活性藥物成分的開(kāi)發(fā)和生產(chǎn)》(2012 年 11 月)所述�。該起始點(diǎn)可能因 API 所用工藝類(lèi)型(如合成����、發(fā)酵、提取�����、純化)而異����。
Surveillance inspections have two inspection options: a full inspection and an abbreviated inspection. For-cause inspections can provide focused coverage, or they can be expanded toabbreviated or full inspections on a case-by-case basis, at OII’s discretion.
監(jiān)督檢查有兩種選擇:全面檢查和簡(jiǎn)化檢查。針對(duì)性檢查可提供聚焦覆蓋�,或 OII 可酌情將其擴(kuò)展為簡(jiǎn)化檢查或全面檢查�����。
(1) Full Inspection Option
全面檢查選項(xiàng)
The full inspection option is a CGMP inspection that provides a broad and in-depth evaluation of the establishment’s conformance with CGMP requirements. A full inspection maychange to an abbreviated inspection with concurrence from OII.
全面檢查是對(duì)機(jī)構(gòu)遵守 CGMP 要求情況進(jìn)行廣泛而深入評(píng)估的 CGMP 檢查����。經(jīng) OII 同意����,全面檢查可改為簡(jiǎn)化檢查。
During the course of a full inspection, coverage in other systems may be needed to verify quality system activities. The full inspection option should include an inspection of at least four systems, one of which must be the quality system.
全面檢查過(guò)程中����,可能需要覆蓋其他系統(tǒng)以驗(yàn)證質(zhì)量系統(tǒng)活動(dòng)。全面檢查應(yīng)包括至少四個(gè)系統(tǒng)的檢查����,其中一個(gè)必須是質(zhì)量系統(tǒng)。
A full inspection is appropriate for the following cases:
全面檢查適用于以下情況:
For an initial inspection of a newly registered establishment. Inspection coverage should include all the systems that are appropriate for the operations.
對(duì)新注冊(cè)機(jī)構(gòu)的首次檢查����。檢查覆蓋范圍應(yīng)包括所有適合其運(yùn)營(yíng)的系統(tǒng)。
When the establishment has a history of fluctuating into and out of compliance. To determine if the establishment meets this criterion, OII should use all of the information at itsdisposal. This information can include inspection results, results of sample analyses, complaints, defects, drug quality reports, field alert reports, adverse event reports, and recalls, inaddition to any compliance actions resulting from these types of information or from past inspections.
機(jī)構(gòu)有合規(guī)狀態(tài)反復(fù)波動(dòng)的歷史����。OII 應(yīng)利用所有可用信息判定機(jī)構(gòu)是否符合此標(biāo)準(zhǔn)���,包括檢查結(jié)果、樣品分析結(jié)果��、投訴��、缺陷�����、藥品質(zhì)量報(bào)告�、現(xiàn)場(chǎng)警報(bào)報(bào)告����、不良事件報(bào)告、召回�����,以及由這些信息或過(guò)去檢查導(dǎo)致的任何合規(guī)措施����。
To evaluate whether important changes have occurred by comparing current operations against the EIR from the last full inspection. The following are typical types of changes that warrant the full inspection option:
通過(guò)將當(dāng)前運(yùn)營(yíng)與上次全面檢查的 EIR 比較,評(píng)估是否發(fā)生重大變化。以下是值得進(jìn)行全面檢查的典型變化:
-Changes that introduce a new potential for cross-contamination through the type of materials that use the same equipment or the type of processing.
通過(guò)使用相同設(shè)備的物料類(lèi)型或工藝類(lèi)型引入新的交叉污染風(fēng)險(xiǎn)的變化�。
-Use of new technology requiring new expertise, significant equipment changes, or new facilities.
使用需要新專(zhuān)業(yè)知識(shí)、重大設(shè)備變更或新設(shè)施的新技術(shù)����。
When OII management or CDER requests this option.
OII 管理層或 CDER 要求此選項(xiàng)時(shí)。
To follow up on a warning letter or other regulatory actions.
跟進(jìn)警告信或其他監(jiān)管措施時(shí)�。
When, at OII’s discretion, a full inspection needs to be conducted based on CGMP findings.
OII 根據(jù) CGMP 檢查結(jié)果酌情決定需要進(jìn)行全面檢查時(shí)。
(2) Abbreviated Inspection Option
簡(jiǎn)化檢查選項(xiàng)
The abbreviated inspection option is a CGMP inspection that efficiently updates the evaluation of and provides documentation for an establishment’s conformance with CGMP requirements. The abbreviated inspection option should include an inspection audit of two to three systems, one of which must be the quality system. OII’s division management should ensure that the optional systems are rotated in successive abbreviated inspections. During the course of an abbreviated inspection, verification of quality system activities may require limited coverage in other systems.
簡(jiǎn)化檢查是高效更新對(duì)機(jī)構(gòu)遵守 CGMP 要求情況的評(píng)估并提供相關(guān)文件的 CGMP 檢查����。簡(jiǎn)化檢查應(yīng)包括對(duì)兩到三個(gè)系統(tǒng)的檢查審計(jì),其中一個(gè)必須是質(zhì)量系統(tǒng)����。OII 的部門(mén)管理層應(yīng)確保在連續(xù)的簡(jiǎn)化檢查中輪換可選系統(tǒng)。簡(jiǎn)化檢查過(guò)程中��,驗(yàn)證質(zhì)量系統(tǒng)活動(dòng)可能需要對(duì)其他系統(tǒng)進(jìn)行有限覆蓋��。
An abbreviated inspection is appropriate when a full inspection is not warranted. This option involves inspecting the manufacturer to: (1) maintain surveillance over the establishment’s manufacturing practices and quality performance; and (2) evaluate whether the establishment is maintaining and improving the CGMP level of assurance for the quality of its APIs. Select the abbreviated inspection option (with OII concurrence) when an establishment has:
當(dāng)無(wú)需全面檢查時(shí)����,簡(jiǎn)化檢查是適當(dāng)?shù)摹T撨x項(xiàng)通過(guò)檢查制造商以:(1)持續(xù)監(jiān)督機(jī)構(gòu)的生產(chǎn)實(shí)踐和質(zhì)量績(jī)效�;(2)評(píng)估機(jī)構(gòu)是否維持并提高 API 質(zhì)量的 CGMP 保證水平��。經(jīng) OII 同意�����,當(dāng)機(jī)構(gòu)符合以下條件時(shí)選擇簡(jiǎn)化檢查:
A record of sustained acceptable compliance history
有持續(xù)可接受的合規(guī)歷史記錄
A strong risk management program
有強(qiáng)有力的風(fēng)險(xiǎn)管理計(jì)劃
A lack of significant marketed API quality defects.
無(wú)重大市售 API 質(zhì)量缺陷
An abbreviated inspection may be changed to a full inspection at OII’s discretion.
OII 可酌情將簡(jiǎn)化檢查改為全面檢查��。
This section provides a complete description of each system and the areas for coverage. Investigators should take the firm’s specific operating conditions, history of previous coverage, and history of CGMP compliance into consideration when selecting systems and determining the relative depth of the audit’s coverage.
本節(jié)提供每個(gè)系統(tǒng)的完整描述及覆蓋領(lǐng)域�。檢查人員選擇系統(tǒng)和確定審計(jì)覆蓋的相對(duì)深度時(shí)�����,應(yīng)考慮企業(yè)的具體運(yùn)營(yíng)條件����、先前覆蓋歷史和 CGMP 合規(guī)歷史��。
The organization and personnel (including appropriate qualifications and training), employed in any given system, will be evaluated as part of that system’s operation. Production, control, or distribution records are required to maintain CGMP compliance, and the recordsselected for review should be included for inspection audit within the context of each of the systems. Inspection of contract companies should include (1) coverage within the system for which the intermediate, API, or service is contracted; and (2) evaluation of their quality system.
任何系統(tǒng)中的組織和人員(包括適當(dāng)?shù)馁Y質(zhì)和培訓(xùn))將作為該系統(tǒng)運(yùn)營(yíng)的一部分進(jìn)行評(píng)估��。生產(chǎn)�����、控制或分銷(xiāo)記錄是維持 CGMP 合規(guī)性所必需的���,所選審查記錄應(yīng)納入每個(gè)系統(tǒng)的檢查審計(jì)范圍��。對(duì)合同公司的檢查應(yīng)包括(1)對(duì)中間體�、API 或所承包服務(wù)所屬系統(tǒng)的覆蓋;(2)對(duì)其質(zhì)量系統(tǒng)的評(píng)估����。
Each of the following system descriptions has a bulleted list of areas. The firm should have written and approved procedures and documentation for each of these areas. Whenever possible, investigators should verify (through observation) if firms are adhering to written procedures. For each system, the areas are not limited to the final API but may also include starting materials and intermediates. All areas under each system should be covered; however, the depth of coverage may vary from the planned inspection strategy depending on inspectional findings.
以下每個(gè)系統(tǒng)描述均包含帶項(xiàng)目符號(hào)的領(lǐng)域列表。企業(yè)應(yīng)為每個(gè)領(lǐng)域制定書(shū)面且經(jīng)批準(zhǔn)的程序和文件����。檢查人員應(yīng)盡可能通過(guò)觀察驗(yàn)證企業(yè)是否遵守書(shū)面程序。每個(gè)系統(tǒng)的領(lǐng)域不僅限于最終 API����,還可能包括起始物料和中間體。每個(gè)系統(tǒng)下的所有領(lǐng)域都應(yīng)被覆蓋����,但覆蓋深度可能根據(jù)檢查結(jié)果與計(jì)劃?rùn)z查策略有所不同。
(1) Quality System
質(zhì)量系統(tǒng)
The quality system is assessed in two phases. In phase one, investigators evaluate whether the quality unit has fulfilled the responsibility to review and approve all procedures related to production, quality control, and quality assurance. Additionally, investigators will evaluate if the quality unit has ensured that the procedures and the associated record keeping systems are adequate for their intended use. In phase two, investigators assess the data and identify any quality problems. This phase may link to other major systems for inspectional coverage.
質(zhì)量系統(tǒng)的評(píng)估分為兩個(gè)階段��。第一階段����,檢查人員評(píng)估質(zhì)量部門(mén)是否履行了審核和批準(zhǔn)所有與生產(chǎn)����、質(zhì)量控制和質(zhì)量保證相關(guān)的程序的職責(zé)��,還將評(píng)估質(zhì)量部門(mén)是否確保這些程序及相關(guān)記錄保存系統(tǒng)適合其預(yù)期用途��。第二階段��,檢查人員評(píng)估數(shù)據(jù)并識(shí)別任何質(zhì)量問(wèn)題����,此階段可能與其他主要系統(tǒng)的檢查覆蓋相關(guān)聯(lián)。
Evaluate each of the areas listed below. If there are additional considerations for the area,they are listed in sub-bullets.
評(píng)估以下每個(gè)領(lǐng)域��。若該領(lǐng)域有其他考慮因素�����,將在子項(xiàng)目符號(hào)中列出����。
Quality oversight of contracted operations and material suppliers
對(duì)承包運(yùn)營(yíng)和物料供應(yīng)商的質(zhì)量監(jiān)督
- An effective monitoring strategy has been implemented; incoming material monitoring, life cycle qualification program, quality agreements, and timely communication mechanisms are implemented.
已實(shí)施有效的監(jiān)控策略��;已實(shí)施來(lái)料監(jiān)控����、生命周期確認(rèn)計(jì)劃�、質(zhì)量協(xié)議和及時(shí)的溝通機(jī)制。
Management oversight of the development, implementation, monitoring, and continual improvement of the quality system
對(duì)質(zhì)量系統(tǒng)的開(kāi)發(fā)����、實(shí)施、監(jiān)控和持續(xù)改進(jìn)的管理監(jiān)督
-Quality risk management and knowledge management are incorporated (e.g., timely and effective communication, appropriate resource allocation, reviews of process performance and API quality).
已納入質(zhì)量風(fēng)險(xiǎn)管理和知識(shí)管理(例如����,及時(shí)有效的溝通、適當(dāng)?shù)馁Y源分配�、過(guò)程性能和 API 質(zhì)量審查)。
lQuality oversight of hazards (e.g., cross-contamination, adulteration, hazardous impurities such as nitrosamines and nitrosating agents)
對(duì)危害(如交叉污染��、摻假����、有害雜質(zhì)(如亞硝胺和亞硝化劑))的質(zhì)量監(jiān)督
-Hazards are documented, identified, evaluated, addressed, communicated, and continually reviewed (as needed) throughout the API’s life cycle.
在 API 的整個(gè)生命周期中��,危害得到記錄�、識(shí)別���、評(píng)估�����、處理����、溝通�,并在需要時(shí)進(jìn)行持續(xù)審查�����。
-Hazardous impurity risks are assessed, and control strategies are implemented to mitigate the risks (e.g., actions to address sources of variability, release testing, reduction or elimination of impurities, cleaning validation); control strategies are reviewed following changes and throughout the API’s life cycle.
對(duì)有害雜質(zhì)風(fēng)險(xiǎn)進(jìn)行了評(píng)估����,并實(shí)施了控制策略以降低風(fēng)險(xiǎn)(如針對(duì)變異性來(lái)源、放行測(cè)試�����、減少或消除雜質(zhì)、清潔驗(yàn)證的行動(dòng))����;在變更后和整個(gè) API 生命周期中對(duì)控制策略進(jìn)行審查。
Adequate staffing to ensure fulfillment of quality unit duties
足夠的人員配置以確保履行質(zhì)量部門(mén)職責(zé)
Periodic quality reviews as described in ICH Q7 ICH
Q7 中所述的定期質(zhì)量審核
-Reviews are complete and conducted at least annually; API quality is reviewed to assess risk and determine the need for changes, such as changes in API specifications, manufacturing, or control procedures; statistical analysis is conducted to identify areas (e.g., trends, patterns, correlations,anomalies) for action and improvement.
審核完整且至少每年進(jìn)行一次;對(duì) API 質(zhì)量進(jìn)行審核以評(píng)估風(fēng)險(xiǎn)并確定是否需要變更(如 API 規(guī)格����、生產(chǎn)或控制程序的變更)�����;進(jìn)行統(tǒng)計(jì)分析以識(shí)別需要采取行動(dòng)和改進(jìn)的領(lǐng)域(如趨勢(shì)���、模式�、相關(guān)性、異常)。
Complaint reviews (quality and medical)
投訴審核(質(zhì)量和醫(yī)療方面)
-Reviews are documented, evaluated, and investigated in a timely manner; corrective action is included when appropriate.
–審核有記錄�、經(jīng)過(guò)評(píng)估并及時(shí)調(diào)查;適當(dāng)時(shí)包括糾正措施�����。
Discrepancies, failures, and critical deviations related to manufacturing and testing
與生產(chǎn)和測(cè)試相關(guān)的差異�����、失敗和關(guān)鍵偏差
-These issues are documented and investigated in a timely manner using scientific evidence to identify the root cause; including corrective actions and preventive actions, and the effectiveness of the corrective and preventative actions is evaluated; investigations are expanded to include any related APIs or materials.
這些問(wèn)題有記錄并及時(shí)調(diào)查��,使用科學(xué)證據(jù)確定根本原因����;包括糾正措施和預(yù)防措施,并評(píng)估糾正和預(yù)防措施的有效性��;調(diào)查范圍擴(kuò)大到包括任何相關(guān)的 API 或物料。
Stability failures
穩(wěn)定性失敗
-Investigation is expanded where warranted; disposition is documented; stability data supports the API retest dates and storage conditions.
在必要時(shí)擴(kuò)大調(diào)查范圍���;處置有記錄;穩(wěn)定性數(shù)據(jù)支持 API 的復(fù)驗(yàn)日期和儲(chǔ)存條件�����。
Change management for the manufacturing of all APIs
所有 API 生產(chǎn)的變更管理
-Changes are documented (with justification), reviewed by subject matter experts, approvedbefore implementation, and evaluated for effectiveness; changes are revalidated, reverified, and requalified as needed; quality risk management is used to evaluate proposed changes for potential risks (e.g., hazardous impurities) and impact on API quality; changes are reported to FDA by the drug application or drug master file (DMF) holder, as appropriate.
–變更有記錄(附有理由),由主題專(zhuān)家審核,在實(shí)施前獲得批準(zhǔn)��,并評(píng)估其有效性;根據(jù)需要對(duì)變更進(jìn)行重新驗(yàn)證、重新確認(rèn)和重新確認(rèn)����;使用質(zhì)量風(fēng)險(xiǎn)管理評(píng)估擬議變更的潛在風(fēng)險(xiǎn)(如有害雜質(zhì))及其對(duì) API 質(zhì)量的影響;由藥品申請(qǐng)或藥品主文件(DMF)持有人酌情向 FDA 報(bào)告變更����。
Reporting of changes for approved drug application products已批準(zhǔn)藥品申請(qǐng)產(chǎn)品的變更報(bào)告
-Changes to established conditions are documented and communicated to the drug application holder, as appropriate. This enables reporting that is compliant with relevant regulationsand consistent with the product life cycle management document in the drug application or recommendations in relevant guidance.
對(duì)既定條件的變更有記錄,并酌情傳達(dá)給藥品申請(qǐng)持有人��。這使得報(bào)告符合相關(guān)法規(guī),并與藥品申請(qǐng)中的產(chǎn)品生命周期管理文件或相關(guān)指南中的建議一致��。
Rejects
拒收
-Investigations are expanded when warranted; corrective actions and preventative actions are implemented, when appropriate.在必要時(shí)擴(kuò)大調(diào)查范圍��;適當(dāng)時(shí)實(shí)施糾正措施和預(yù)防措施。
Quality oversight system for the release or rejection of raw materials
原料放行或拒收的質(zhì)量監(jiān)督系統(tǒng)
Batches manufactured since the last inspection to evaluate any rejections or conversions (e.g., from drug to nondrug use) because of processing problems
自上次檢查以來(lái)生產(chǎn)的批次����,以評(píng)估因加工問(wèn)題導(dǎo)致的任何拒收或轉(zhuǎn)換(如從藥品用途轉(zhuǎn)為非藥品用途)
Recalls (including any attempt to recover distributed API not meeting its specifications or purported quality)
召回(包括任何回收不符合其規(guī)格或聲稱(chēng)質(zhì)量的已分銷(xiāo) API 的嘗試)
-The cause was determined; corrective actions were taken.
已確定原因;已采取糾正措施���。
Validation
驗(yàn)證
-The statuses of validation and revalidation activities (e.g., computer, manufacturing process, laboratory methods) are documented (e.g., reviews and approvals of validation protocols and reports).
–驗(yàn)證和再驗(yàn)證活動(dòng)(如計(jì)算機(jī)����、生產(chǎn)過(guò)程、實(shí)驗(yàn)室方法)的狀態(tài)有記錄(如驗(yàn)證方案和報(bào)告的審核和批準(zhǔn))�。
Contemporaneous and complete documentation
同期且完整的文件記錄
CGMP training and qualification for employees on a continuing basis and with sufficient frequency
持續(xù)且足夠頻繁地對(duì)員工進(jìn)行 CGMP 培訓(xùn)和資質(zhì)認(rèn)定
-Training includes coverage of quality functions, risk management, and the specific CGMP operations assigned to individual employees.
–培訓(xùn)包括質(zhì)量職能�����、風(fēng)險(xiǎn)管理以及分配給每位員工的特定 CGMP 操作的內(nèi)容�。
Programs for the ongoing monitoring of process performance and API quality throughout the API’s life cycle
在整個(gè) API 生命周期中對(duì)過(guò)程性能和 API 質(zhì)量進(jìn)行持續(xù)監(jiān)控的程序
-Significant issues are escalated to senior management.
重大問(wèn)題上報(bào)給高級(jí)管理層。
Reprocess and rework
返工和再加工
-Evaluation is conducted; approval is documented; impact on validation and stability is assessed.
進(jìn)行評(píng)估��;批準(zhǔn)有記錄;評(píng)估對(duì)驗(yàn)證和穩(wěn)定性的影響��。
Returns and salvages
退貨和回收
-Assessment is conducted; investigation is expanded where warranted; disposition is completed.
進(jìn)行評(píng)估�����;在必要時(shí)擴(kuò)大調(diào)查范圍����;完成處置。
(2)Facilities and Equipment System
設(shè)施與設(shè)備系統(tǒng)
(a) Facilities
設(shè)施
Evaluate each of the areas listed below. If there are additional considerations for the area they are listed in sub-bullets.
評(píng)估以下每個(gè)領(lǐng)域�。若該領(lǐng)域有其他考慮因素,將在子項(xiàng)目符號(hào)中列出�。
Responsible personnel’s oversight of the facility infrastructure and the suitability of manufacturing operations
負(fù)責(zé)人員對(duì)設(shè)施基礎(chǔ)設(shè)施和生產(chǎn)操作適用性的監(jiān)督
Change management system for implementing changes in the facility
用于實(shí)施設(shè)施變更的變更管理系統(tǒng)
Facility layout, material flow, and personnel flow that prevent cross-contamination, including cross-contamination from processing of nondrug materials
防止交叉污染的設(shè)施布局、物料流和人員流,包括來(lái)自非藥品物料加工的交叉污染
Complete and comprehensive separation of the manufacturing operations for highly sensitizing agents (e.g., penicillin, beta-lactams, steroids, hormones, and cytotoxics)
高致敏性物質(zhì)(如青霉素��、β- 內(nèi)酰胺類(lèi)�����、類(lèi)固醇��、激素和細(xì)胞毒素)生產(chǎn)操作的完全和全面分離
Qualified and appropriately monitored utilities (e.g., steam, gas, compressed air, heating, ventilation, air conditioning)
經(jīng)過(guò)確認(rèn)且適當(dāng)監(jiān)控的公用設(shè)施(如蒸汽��、氣體、壓縮空氣�����、供暖��、通風(fēng)�����、空調(diào))
Lighting, potable water, washing and toilet facilities, and sewage and refuse disposal
照明����、飲用水、洗滌和衛(wèi)生間設(shè)施以及污水和垃圾處理
Cleaning and maintenance that potentially affect API quality
可能影響 API 質(zhì)量的清潔和維護(hù)
Sanitation of the facility and use of rodenticides, fungicides, insecticides, and cleaning andsanitizing agents
設(shè)施的衛(wèi)生以及滅鼠劑��、殺真菌劑、殺蟲(chóng)劑以及清潔和消毒劑的使用
Training and qualification of personnel
人員的培訓(xùn)和資質(zhì)認(rèn)定
(b) Equipment
設(shè)備
Evaluate each of the areas listed below. If there are additional considerations for the area, they are listed in sub-bullets.
評(píng)估以下每個(gè)領(lǐng)域�����。若該領(lǐng)域有其他考慮因素��,將在子項(xiàng)目符號(hào)中列出��。
Equipment installation
設(shè)備安裝
Equipment is operational; performance is qualified, when appropriate.
設(shè)備可運(yùn)行�;性能已適當(dāng)確認(rèn)����。
Adequate equipment design, size, and location
足夠的設(shè)備設(shè)計(jì)、尺寸和位置
Controls to prevent contamination, including appropriate design provisions to ensure separation from pesticides, other toxic materials, or nondrug chemicals
防止污染的控制措施��,包括確保與農(nóng)藥��、其他有毒物質(zhì)或非藥品化學(xué)品分離的適當(dāng)設(shè)計(jì)規(guī)定
Equipment surfaces that are not reactive, additive, or absorptive in a way that could alter material quality
設(shè)備表面不會(huì)以可能改變物料質(zhì)量的方式發(fā)生反應(yīng)�、添加物質(zhì)或吸收物質(zhì)
Appropriate identification of equipment (e.g., reactors, storage containers) and permanently installed processing lines
設(shè)備(如反應(yīng)器、儲(chǔ)存容器)和永久安裝的加工管線(xiàn)的適當(dāng)標(biāo)識(shí)
Prevention of contact between substances associated with the operation of equipment (e.g.,lubricants, heating fluids, coolants) and starting materials, intermediates, final APIs, and containers
防止與設(shè)備操作相關(guān)的物質(zhì)(如潤(rùn)滑劑�、加熱流體�����、冷卻液)與起始物料�、中間體、最終 API 和容器接觸
Cleaning procedures and cleaning validation and sanitization studies to verify that residues,microbial contamination, and, when appropriate, endotoxin contamination are brought belowscientifically appropriate levels
清潔程序以及清潔驗(yàn)證和消毒研究���,以驗(yàn)證殘留物�����、微生物污染以及適當(dāng)時(shí)的內(nèi)毒素污染降至科學(xué)上適當(dāng)?shù)乃揭韵?/span>
Calibrations that use standards that can be traced to certified standards (e.g., National Institute of Standards and Technology, United States Pharmacopeia (USP))
使用可追溯至經(jīng)認(rèn)證標(biāo)準(zhǔn)(如美國(guó)國(guó)家標(biāo)準(zhǔn)與技術(shù)研究院��、美國(guó)藥典(USP))的標(biāo)準(zhǔn)進(jìn)行校準(zhǔn)
Qualification, calibration, and maintenance of storage equipment (e.g., refrigerators, freezers) to ensure that materials (e.g., standards, raw materials, reagents) are stored under appropriate conditions
儲(chǔ)存設(shè)備(如冰箱、冰柜)的確認(rèn)�����、校準(zhǔn)和維護(hù)���,以確保物料(如標(biāo)準(zhǔn)品����、原料�����、試劑)在適當(dāng)條件下儲(chǔ)存
Equipment (including computers) qualification, validation, calibration, maintenance, and security
設(shè)備(包括計(jì)算機(jī))的確認(rèn)、驗(yàn)證����、校準(zhǔn)、維護(hù)和安全
Control system for implementing changes to equipment
用于實(shí)施設(shè)備變更的控制系統(tǒng)
Documentation of any discrepancies (critical discrepancy investigations are covered under the quality system)
任何差異的文件記錄(關(guān)鍵差異調(diào)查在質(zhì)量系統(tǒng)下涵蓋)
Training and qualification of personnel
人員的培訓(xùn)和資質(zhì)認(rèn)定
(3) Materials System
物料系統(tǒng)
Evaluate each of the areas listed below. If there are additional considerations for the area,they are listed in sub-bullets.
評(píng)估以下每個(gè)領(lǐng)域�����。若該領(lǐng)域有其他考慮因素����,將在子項(xiàng)目符號(hào)中列出。
Training and qualification of personnel
人員的培訓(xùn)和資質(zhì)認(rèn)定
Identification of starting materials and containers
起始物料和容器的標(biāo)識(shí)
Storage conditions
儲(chǔ)存條件
Quarantine of all materials and APIs (including reprocessed materials) until they are testedor examined and released
所有物料和 API(包括返工物料)在測(cè)試或檢查并放行前的待驗(yàn)
Collection of representative samples for testing or examination
收集代表性樣品進(jìn)行測(cè)試或檢查
-Appropriate means are used; comparisons are against appropriate specifications.使用適當(dāng)?shù)姆椒?��;與適當(dāng)?shù)囊?guī)格進(jìn)行比較���。
A system for auditing and monitoring the suppliers of critical materials (e.g., raw materials, starting materials, intermediates) and containers
對(duì)關(guān)鍵物料(如原料、起始物料�����、中間體)和容器的供應(yīng)商進(jìn)行審計(jì)和監(jiān)控的系統(tǒng)
Rejected materials
拒收的物料
-Decisions to keep or reject materials that do not meet acceptance requirements (e.g., starting materials, intermediates, containers) are documented and justified; rejected materials arepromptly quarantined and disposed.
對(duì)不符合驗(yàn)收要求的物料(如起始物料���、中間體��、容器)的保留或拒收決定有記錄和理由��;拒收的物料及時(shí)隔離和處置��。
Appropriate retesting or reexamination of starting materials, intermediates, and containers
起始物料�����、中間體和容器的適當(dāng)重新測(cè)試或重新檢查
First in, first out use of materials and containers
物料和容器的先進(jìn)先出使用
Suitability of process water used to manufacture APIs, including the water system design, maintenance, validation, and operation, as appropriate
用于生產(chǎn) API 的工藝用水的適用性����,包括水系統(tǒng)的設(shè)計(jì)、維護(hù)�����、驗(yàn)證和操作(視情況而定)
Suitability of process gas used in the manufacture of APIs (e.g., gas use to sparge a reactor), including the gas system design, maintenance, validation, and operation, as appropriate
用于 API 生產(chǎn)的工藝氣體(如用于反應(yīng)器鼓泡的氣體)的適用性���,包括氣體系統(tǒng)的設(shè)計(jì)、維護(hù)��、驗(yàn)證和操作(視情況而定)
Containers and closures that are not additive, reactive, or absorptive
非添加性�、非反應(yīng)性和非吸收性的容器和密封件
Change management system for implementing changes in material handling operations
用于實(shí)施物料處理操作變更的變更管理系統(tǒng)
-Changes in the supply chain for critical materials and containers are thoroughly evaluated,approved, and documented to ensure that they are unlikely to pose an adverse risk to APIquality.
– 對(duì)關(guān)鍵物料和容器供應(yīng)鏈的變更進(jìn)行徹底評(píng)估、批準(zhǔn)和記錄�,以確保它們不太可能對(duì) API 質(zhì)量造成不利風(fēng)險(xiǎn)��。
Qualification, validation, and security of computerized or automated processes
計(jì)算機(jī)化或自動(dòng)化過(guò)程的確認(rèn)��、驗(yàn)證和安全
Distribution records by batch for all materials (e.g., the finished API, intermediates), including records for exported materials
所有物料(如成品 API���、中間體)的批次分銷(xiāo)記錄,包括出口物料的記錄
Documentation of any discrepancies (critical discrepancy investigations are covered under the quality system)
任何差異的文件記錄(關(guān)鍵差異調(diào)查在質(zhì)量系統(tǒng)下涵蓋)
Risk management program for starting materials, intermediates, and containers
起始物料����、中間體和容器的風(fēng)險(xiǎn)管理計(jì)劃
-Unacceptable hazards (e.g., impurities) are addressed; risks are assessed, as needed, throughout the API’s life cycle.
解決不可接受的危害(如雜質(zhì));在 API 的整個(gè)生命周期中根據(jù)需要評(píng)估風(fēng)險(xiǎn)����。
(4) Production System
生產(chǎn)系統(tǒng)
Evaluate each of the areas listed below. If there are additional considerations for the area,they are listed in sub-bullets.
評(píng)估以下每個(gè)領(lǐng)域。若該領(lǐng)域有其他考慮因素�����,將在子項(xiàng)目符號(hào)中列出�����。
Training and qualification of personnel
人員的培訓(xùn)和資質(zhì)認(rèn)定
Establishment of, adherence to, and documented performance of approved manufacturing procedures
制定�����、遵守經(jīng)批準(zhǔn)的生產(chǎn)程序并記錄其執(zhí)行情況
Controls for critical activities and operations
對(duì)關(guān)鍵活動(dòng)和操作的控制
Documentation and investigation of critical deviations
關(guān)鍵偏差的文件記錄和調(diào)查
Comparison of actual yields with expected yields at designated steps
在指定步驟將實(shí)際收率與預(yù)期收率進(jìn)行比較
Process validation program that ensures a state of control across the life cycle (i.e., process design, process qualification, and continued process verification stages)
確保整個(gè)生命周期(即過(guò)程設(shè)計(jì)、過(guò)程確認(rèn)和持續(xù)過(guò)程驗(yàn)證階段)處于受控狀態(tài)的工藝驗(yàn)證計(jì)劃
Establishment of adequate control steps to ensure that APIs are suitable and safe for theirintended use
制定充分的控制步驟�,確保 API 適合并安全用于其預(yù)期用途
-For APIs intended to be used in parenteral dosage forms, pyrogenic (e.g., endotoxin) contamination can be reduced or prevented through the use of appropriate water quality, inputmaterials, and process steps.
對(duì)于擬用于注射劑型的 API,可通過(guò)使用適當(dāng)?shù)乃|(zhì)����、輸入物料和工藝步驟來(lái)減少或防止熱原(如內(nèi)毒素)污染。
Established time limits for completion of phases of production, when appropriate
適當(dāng)時(shí)��,規(guī)定生產(chǎn)各階段的完成時(shí)限
Appropriate identification of major equipment used in the production of intermediates and the API
用于生產(chǎn)中間體和 API 的主要設(shè)備的適當(dāng)標(biāo)識(shí)
Justification and consistency of intermediate specifications and API specifications
中間體規(guī)格和 API 規(guī)格的合理性和一致性
Implementation and documentation of process controls, testing, and examinations (e.g., pH,temperature, purity, actual yields, clarity)
過(guò)程控制����、測(cè)試和檢查(如 pH 值、溫度����、純度、實(shí)際收率��、澄清度)的實(shí)施和文件記錄
In-process sampling that is conducted using procedures designed to prevent contamination of the sampled material
按照旨在防止被采樣物料污染的程序進(jìn)行過(guò)程中采樣
Recovery (e.g., from mother liquor or filtrates) of reactants
反應(yīng)物的回收(如從母液或?yàn)V液中)
-Approved procedures and recovered materials meet specifications that are suitable for their intended use.
經(jīng)批準(zhǔn)的程序和回收的物料符合適合其預(yù)期用途的規(guī)格���。
Recovery of solvents
溶劑的回收
-Recovered solvents should be used only in the same step or in an earlier step (if there is sufficient purification) of the same processes from which they were collected.
回收的溶劑僅可用于其收集來(lái)源的相同工藝的相同步驟或更早步驟(如果經(jīng)過(guò)充分純化)。
Precautions to prevent or minimize the potential for cross-contamination when APIs are micronized on multiuse equipment當(dāng) API 在多用途設(shè)備上進(jìn)行微粉化時(shí)��,采取預(yù)防措施防止或最大限度地減少交叉污染的可能性
Validation and security of computerized or automated processes
計(jì)算機(jī)化或自動(dòng)化流程的驗(yàn)證和安全
Ongoing statistical evaluations (e.g., batch control data, periodic capability analysis) to identify processes that exhibit high variability and trigger needed improvements
持續(xù)的統(tǒng)計(jì)評(píng)估(如批次控制數(shù)據(jù)、定期能力分析)��,以識(shí)別表現(xiàn)出高變異性的過(guò)程并觸發(fā)所需的改進(jìn)
Change management system for production changes, including evaluation of the need for additional validation studies
生產(chǎn)變更的變更管理系統(tǒng)�����,包括評(píng)估是否需要額外的驗(yàn)證研究
Master batch production and control records that include instructions and processes of appropriate specificity that enable production operators to reproducibly execute the same manufacturing processes each time the API is produced
主批次生產(chǎn)和控制記錄��,包括具有適當(dāng)特異性的說(shuō)明和過(guò)程��,使生產(chǎn)操作員能夠每次生產(chǎn) API 時(shí)可重復(fù)地執(zhí)行相同的生產(chǎn)過(guò)程
Batch production and control records
批次生產(chǎn)和控制記錄
Contemporaneous and complete documentation of batch production
批次生產(chǎn)的同期且完整的文件記錄
Documentation of any discrepancies (critical discrepancy investigations are covered under the quality system)
任何差異的文件記錄(關(guān)鍵差異調(diào)查在質(zhì)量系統(tǒng)下涵蓋)
Establishment of effective control strategies for manufacturing operations that may potentially pose a risk of forming hazardous impurities
為可能存在形成有害雜質(zhì)風(fēng)險(xiǎn)的生產(chǎn)操作制定有效的控制策略
(5) Packaging and Labeling System
包裝與貼標(biāo)簽系統(tǒng)
Evaluate each of the areas listed below. If there are additional considerations for the area,they are listed in sub-bullets.
評(píng)估以下每個(gè)領(lǐng)域�。若該領(lǐng)域有其他考慮因素,將在子項(xiàng)目符號(hào)中列出����。
Training and qualification of personnel
人員的培訓(xùn)和資質(zhì)認(rèn)定
Acceptance operations for packaging and labeling materials
包裝和貼標(biāo)簽物料的驗(yàn)收操作
Establishment of, adherence to, and documented performance of approved packaging and labeling procedures
制定、遵守經(jīng)批準(zhǔn)的包裝和貼標(biāo)簽程序并記錄其執(zhí)行情況
Change management system for implementing changes in packaging and labeling operations
用于實(shí)施包裝和貼標(biāo)簽操作變更的變更管理系統(tǒng)
Adequate storage for labels and labeling, both approved and returned after issued
已批準(zhǔn)和發(fā)放后退回的標(biāo)簽和貼標(biāo)簽的充分儲(chǔ)存
Control of labels with similar sizes, shapes, and colors that are used for different APIs
用于不同 API 的具有相似尺寸�����、形狀和顏色的標(biāo)簽的控制
Adequate packaging records that include specimens of all labels used充分的包裝記錄�,包括所用所有標(biāo)簽的樣本
Control of issuance of labeling, examination of issued labels, and reconciliation of used labels標(biāo)簽發(fā)放的控制、所發(fā)放標(biāo)簽的檢查以及所用標(biāo)簽的核對(duì)
Examination of the labeled finished APIs
對(duì)貼標(biāo)簽的成品API 的檢查
Adequate inspection (i.e., proofing) of incoming labeling
對(duì)收到的貼標(biāo)簽的充分檢查(即校對(duì))
Use of lot numbers and destruction of excess labeling bearing lot or control numbers
批號(hào)的使用以及銷(xiāo)毀帶有批號(hào)或控制號(hào)的多余標(biāo)簽
Adequate separation and controls when more than one batch is labeled at a time
當(dāng)同時(shí)對(duì)多個(gè)批次進(jìn)行貼標(biāo)簽時(shí)的充分分離和控制
Adequate expiration or retest dates on the label
標(biāo)簽上充分的有效期或復(fù)驗(yàn)日期
Validation of packaging and labeling operations including validation and security of computerized processes
包裝和貼標(biāo)簽操作的驗(yàn)證����,包括計(jì)算機(jī)化過(guò)程的驗(yàn)證和安全
Documentation of any discrepancies (critical discrepancy investigations are covered under the quality system)
任何差異的文件記錄(關(guān)鍵差異調(diào)查在質(zhì)量系統(tǒng)下涵蓋)
Repackaged APIs and intermediates
重新包裝的 API 和中間體
-The name of and information about the original API or intermediate manufacturer, repacker, and relabeler are provided.提供原始 API 或中間體制造商����、重新包裝商和重新貼標(biāo)簽商的名稱(chēng)和信息�����。
(6) Laboratory Control System
實(shí)驗(yàn)室控制系統(tǒng)
Evaluate each of the areas listed below. If there are additional considerations for the area,they are listed in sub-bullets.
評(píng)估以下每個(gè)領(lǐng)域�。若該領(lǐng)域有其他考慮因素,將在子項(xiàng)目符號(hào)中列出����。
Training and qualification of personnel
人員的培訓(xùn)和資質(zhì)認(rèn)定
Adequate staffing for laboratory operations
實(shí)驗(yàn)室操作的充分人員配置
Adequate equipment and facility for the intended use
適合預(yù)期用途的充分設(shè)備和設(shè)施
Calibration and maintenance programs for analytical instruments and equipment
分析儀器和設(shè)備的校準(zhǔn)和維護(hù)計(jì)劃
Validation and security of computerized or automated processes
計(jì)算機(jī)化或自動(dòng)化流程的驗(yàn)證和安全
Source and purity of reference standards
參考標(biāo)準(zhǔn)品的來(lái)源和純度
Assays and tests are used to establish equivalency to current official reference standards, as appropriate.測(cè)定和測(cè)試用于確定與當(dāng)前官方參考標(biāo)準(zhǔn)品的等效性(視情況而定)。
System suitability tests for chromatographic systems
色譜系統(tǒng)的系統(tǒng)適用性測(cè)試
Specifications, standards, and representative sampling plans
規(guī)格��、標(biāo)準(zhǔn)和代表性采樣計(jì)劃
Validation or verification of analytical methods, including suitability of microbiological test methods
分析方法的驗(yàn)證或確認(rèn)����,包括微生物測(cè)試方法的適用性
Adequate test methods for establishing complete impurity profiles for each API productionprocess
用于為每個(gè) API 生產(chǎn)過(guò)程建立完整雜質(zhì)概況的充分測(cè)試方法
Note that impurity profiles are often process-related.
注意雜質(zhì)概況通常與過(guò)程相關(guān)。
Reassessment of impurity profiles after changes that could affect the impurity profile (e.g.,new sources of raw materials or reagents)
在可能影響雜質(zhì)概況的變更后(如原料或試劑的新來(lái)源)重新評(píng)估雜質(zhì)概況
Change management strategy for hazardous impurities if any hazardous impurities have been identified: (1) in any starting material, intermediate, container, or the API; or (2) as a degradant during the API’s life cycle
如果在任何起始物料�����、中間體���、容器或 API 中已識(shí)別出任何有害雜質(zhì)��,或在 API 的生命周期中作為降解產(chǎn)物識(shí)別出任何有害雜質(zhì)���,則針對(duì)有害雜質(zhì)的變更管理策略
Change management system for implementing changes in laboratory operations
用于實(shí)施實(shí)驗(yàn)室操作變更的變更管理系統(tǒng)
Required testing of the correct samples using the approved or filed methods or equivalent methods
使用批準(zhǔn)或備案的方法或等效方法對(duì)正確的樣品進(jìn)行所需的測(cè)試
Documentation of any discrepancies (critical discrepancy investigations are covered under the quality system)
任何差異的文件記錄(關(guān)鍵差異調(diào)查在質(zhì)量系統(tǒng)下涵蓋)
Complete analytical records from all tests and summaries of results
所有測(cè)試的完整分析記錄和結(jié)果摘要
Contemporaneous and complete laboratory records
同期且完整的實(shí)驗(yàn)室記錄
Quality and retention of raw data (e.g., chromatograms, spectra)
原始數(shù)據(jù)(如色譜圖、光譜)的質(zhì)量和保留
Correlation of result summaries to raw data; presence and disposition of unused data
結(jié)果摘要與原始數(shù)據(jù)的相關(guān)性����;未使用數(shù)據(jù)的存在和處置
Adherence to an adequate out of specification procedure that includes timely completion ofinvestigations遵守充分的超出規(guī)格程序,包括及時(shí)完成調(diào)查
Adequate reserve samples and documented examination of reserve samples
充分的留樣和留樣的有記錄檢查
Stability testing program, including demonstration that the analytical methods are stability-indicating
穩(wěn)定性測(cè)試計(jì)劃��,包括證明分析方法具有穩(wěn)定性能力
These procedures are in addition to those in the IOM.
這些程序是對(duì)《調(diào)查操作手冊(cè)》中程序的補(bǔ)充�����。
1.Select two or more systems for inspection coverage, as appropriate (see Part III.1.A).
酌情選擇兩個(gè)或多個(gè)系統(tǒng)進(jìn)行檢查覆蓋(見(jiàn)第三部分第 1.A 節(jié))�。
2.If APIs are not specified in the assignment, select significant APIs for inspection coverage. APIs are considered significant based on risk. Significant APIs include: (1) APIs that broadly use all of the systems in the firm or use special manufacturing features (e.g., complex chemical synthesis); and (2) APIs that are highly sensitizing materials, infectious materials, or new chemical entities made under approved drug applications. Review the firm’s inspection history, compliance history, DMF, or drug application files.
如果任務(wù)中未指定 API,則選擇重要的 API 進(jìn)行檢查覆蓋����。API 的重要性基于風(fēng)險(xiǎn)確定。重要 API 包括:(1)廣泛使用企業(yè)所有系統(tǒng)或具有特殊生產(chǎn)特征(如復(fù)雜化學(xué)合成)的 API��;(2)高致敏性材料����、傳染性材料或根據(jù)已批準(zhǔn)藥品申請(qǐng)生產(chǎn)的新化學(xué)實(shí)體����。審查企業(yè)的檢查歷史�、合規(guī)歷史、DMF 或藥品申請(qǐng)文件�。
3.If CDER or OII request a CDER staff member to be a member of the inspection team,the lead investigator should brief them on the intended inspection strategy and explain their supporting role and responsibilities for the inspection. The lead investigator should consult with OPQ assessors on any specific drug application chemistry, manufacturing, or control issues (whether premarket or postmarket) that will be covered during the inspection.
如果 CDER 或 OII 要求 CDER 工作人員加入檢查團(tuán)隊(duì)�����,首席檢查人員應(yīng)向他們簡(jiǎn)要介紹預(yù)期的檢查策略�,并說(shuō)明他們?cè)跈z查中的支持角色和職責(zé)。首席檢查人員應(yīng)就檢查期間將涵蓋的任何特定藥品申請(qǐng)的化學(xué)����、生產(chǎn)和控制問(wèn)題(無(wú)論是上市前還是上市后)咨詢(xún) OPQ 評(píng)估員。
4.Review the impurity profile for each API production process that will be covered during the inspection. If an application or DMF was submitted, compare the firm’s impurity profiles to the impurity profiles submitted.
審查將在檢查期間涵蓋的每個(gè) API 生產(chǎn)過(guò)程的雜質(zhì)概況����。如果已提交申請(qǐng)或 DMF,將企業(yè)的雜質(zhì)概況與所提交的雜質(zhì)概況進(jìn)行比較�。
5.For the APIs that will be inspected, verify conformity to any compendial monographs, as appropriate.
對(duì)于將接受檢查的 API,酌情驗(yàn)證其是否符合任何藥典專(zhuān)著��。
6.Before or during the inspection, determine if the firm has made process changes by comparing current operations against the EIR for the previous inspection. Also compare the current operations with those described in the DMF or the drug application to determine whether the firm is complying with the postapproval chemistry, manufacturing, and controls commitments that they made to FDA. The following are examples of changes that would warrant extensive coverage during the inspection:
在檢查前或檢查期間�����,通過(guò)將當(dāng)前運(yùn)營(yíng)與上一次檢查的 EIR 進(jìn)行比較�����,確定企業(yè)是否進(jìn)行了工藝變更��。還將當(dāng)前運(yùn)營(yíng)與 DMF 或藥品申請(qǐng)中描述的運(yùn)營(yíng)進(jìn)行比較�����,以確定企業(yè)是否遵守其向 FDA 做出的上市后化學(xué)�����、生產(chǎn)和控制承諾�����。以下是在檢查期間值得廣泛覆蓋的變更示例:
a.API production process changes or product-type line changes that include processing of several APIs of varying toxicity in common equipment and/or facilities. These changes could introduce new potential cross-contamination.
API 生產(chǎn)工藝變更或產(chǎn)品線(xiàn)變更�����,包括在共同設(shè)備和 / 或設(shè)施中加工多種不同毒性的 API�����。這些變更可能引入新的潛在交叉污染。
b.New technology that requires new expertise, significantly new equipment, or new facilities.
需要新專(zhuān)業(yè)知識(shí)���、顯著新設(shè)備或新設(shè)施的新技術(shù)�。
c.Changes (particularly those that are not referenced in the DMF or drug application) in starting materials, intermediates, equipment, facilities, support systems, processing steps, packaging materials, or computer software.
起始物料����、中間體、設(shè)備�����、設(shè)施�����、支持系統(tǒng)�����、加工步驟����、包裝材料或計(jì)算機(jī)軟件的變更(特別是那些未在 DMF 或藥品申請(qǐng)中提及的變更)�。
7.For foreign firms, obtain file information from the appropriate CDER assessment division or compliance unit.Investigators may also request background information about the site directly from the U.S. Agent before the inspection.
對(duì)于外國(guó)企業(yè)�,從適當(dāng)?shù)?CDER 評(píng)估部門(mén)或合規(guī)單位獲取文件信息。檢查人員還可在檢查前直接向美國(guó)代理請(qǐng)求該場(chǎng)地的背景信息����。
2. Reporting
報(bào)告
Investigators should describe their inspection coverage and findings in the EIR. Investigators should include a sufficient level of detail for further FDA evaluation of the firm’s state of control and conformance to CGMP requirements. ICH Q7 may be used as a guideline to describe coverage, findings, and deficiencies. However, investigators should not reference specific sections of ICH Q7 in the Form FDA 483 observations or in the EIR. If an investigator believes that a particular practice consistent with ICH Q7 is deficient, the investigator or division should consult with CDER’s Office of Manufacturing Quality (OMQ) before making an observation that conflicts with ICH Q7.
檢查人員應(yīng)在 EIR 中描述其檢查覆蓋范圍和發(fā)現(xiàn)�����。檢查人員應(yīng)提供足夠詳細(xì)的信息��,供 FDA 進(jìn)一步評(píng)估企業(yè)的受控狀態(tài)和對(duì) CGMP 要求的符合性�����。ICH Q7 可用作描述覆蓋范圍�、發(fā)現(xiàn)和缺陷的指南。然而��,檢查人員不應(yīng)在 FDA 483 表觀察結(jié)果或 EIR 中引用 ICH Q7 的具體章節(jié)�����。如果檢查人員認(rèn)為符合 ICH Q7 的特定做法存在缺陷,檢查人員或部門(mén)應(yīng)在做出與 ICH Q7 沖突的觀察結(jié)果之前咨詢(xún) CDER 的生產(chǎn)質(zhì)量辦公室(OMQ)����。
The Form FDA 483, if issued, should include sections for each of the covered systems. In addition to the IOM format and information reporting requirements, all EIRs for API manufacturers must include:
如發(fā)布 FDA 483 表,應(yīng)包含每個(gè)被覆蓋系統(tǒng)的部分��。除《調(diào)查操作手冊(cè)》的格式和信息報(bào)告要求外��,所有API 制造商的 EIR 必須包括:
1.A list of APIs manufactured (or categories of APIs if there are several APIs) along with the general manufacturing process for each API (e.g., chemical synthesis, fermentation, extraction of botanical material).
所生產(chǎn) API 的列表(或 API 類(lèi)別����,如果有多個(gè) API)以及每種 API 的一般生產(chǎn)過(guò)程(如化學(xué)合成、發(fā)酵�����、植物材料提?���。?/span>
2.An explanation for the APIs selected for coverage.
對(duì)所選 API 進(jìn)行覆蓋的解釋
3.For foreign API manufacturers, the U.S. Agent’s name, title, complete mailing address, telephone number, and fax number or email address.
對(duì)于外國(guó) API 制造商����,美國(guó)代理的姓名、頭銜����、完整郵寄地址��、電話(huà)號(hào)碼以及傳真號(hào)碼或電子郵件地址�����。
4.For foreign API manufacturers, a report of all APIs imported into the United States in the last two years, their consignees (including in-country intermediary repackers, relabelers, wholesalers, distributors, and shippers that import directly in the U.S.), and an estimate of the frequency and quantity of shipments to these consignees.
對(duì)于外國(guó) API 制造商����,過(guò)去兩年中所有進(jìn)口到美國(guó)的 API 的報(bào)告��、其收貨人(包括國(guó)內(nèi)中間重新包裝商�����、重新貼標(biāo)簽商��、批發(fā)商�、分銷(xiāo)商和直接進(jìn)口的托運(yùn)人)����,以及對(duì)這些收貨人的發(fā)貨頻率和數(shù)量的估計(jì)。
4. A description of each of the systems selected for coverage (i.e., areas, processes, and operations), what was covered, who was interviewed, and what manufacturing activities were taking place during the inspection.
對(duì)每個(gè)所選覆蓋系統(tǒng)的描述(即領(lǐng)域���、過(guò)程和操作)�、所覆蓋的內(nèi)容、訪談的人員以及檢查期間正在進(jìn)行的生產(chǎn)活動(dòng)���。
5.Any significant changes to a firm’s packaging, labeling, product line, or processes, particularly changes that are not properly filed, submitted, or reported in a DMF or drug application (including changes to APIs intended for use in compounding).企業(yè)包裝����、貼標(biāo)簽�、產(chǎn)品線(xiàn)或過(guò)程的任何重大變更,特別是那些未在 DMF 或藥品申請(qǐng)中適當(dāng)歸檔�����、提交或報(bào)告的變更(包括擬用于配制的 API 的變更)�����。
Investigators should instruct management to submit an electronic response to a Form FDA 483 with appropriate documentation via email to CDER-OC-OMQ-Domestic483Response@fda.hhs.gov or CDER-OC-OMQ-International483Response@fda.hhs.gov.
檢查人員應(yīng)指示管理層通過(guò)電子郵件向CDER-OC-OMQ-Domestic483Response@fda.hhs.gov 或CDER-OC-OMQ-International483Response@fda.hhs.gov 提交對(duì)FDA 483表的電子回復(fù)及適當(dāng)文件��。
For human drug inspections that include CGMP and preapproval inspectional coverage, in addition to the address above, include: CDERPAIprogram@fda.hhs.gov.
對(duì)于包括CGMP和批準(zhǔn)前檢查覆蓋的人用藥品檢查 �, 除上述地址外��,還應(yīng)發(fā)送至:CDERPAIprogram@fda.hhs.gov�����。
PART IV – ANALYTICAL
第四部分-分析
API samples that the investigator collects for quality evaluation should be submitted to theappropriate servicing laboratory. Please email the Office of the Chief Scientist (OCS)/Office of Analytical and Regulatory Laboratories (OARL) at OCOCSOARLProgramCoordinators@fda.hhs.gov to request servicing laboratories for chemical and microbiological testing. In the request, include the API description, lot numbers to be tested, analyses required, and the reason for sample collection. Servicing laboratories will be selected based on specialization, technology and testing expertise, and capacity. However, it should be noted that physical API samples are not required to support regulatory or administrative action against a violative firm or drug.
檢查人員為質(zhì)量評(píng)估而收集的 API 樣品應(yīng)提交給適當(dāng)?shù)姆?wù)實(shí)驗(yàn)室。請(qǐng)通過(guò)電子郵件向首席科學(xué)家辦公室( OCS ) / 分 析 和 監(jiān) 管 實(shí) 驗(yàn) 室 辦 公 室 ( OARL ) 發(fā) 送 請(qǐng) 求 ���, 郵 箱 地 址 為 OCOCSOARLProgramCoordinators@fda.hhs.gov�����,請(qǐng)求提供用于化學(xué)和微生物測(cè)試的服務(wù)實(shí)驗(yàn)室���。在請(qǐng)求中,應(yīng)包括 API 描述���、待測(cè)試的批號(hào)、所需的分析以及樣品收集的原因�。服務(wù)實(shí)驗(yàn)室的選擇將基于專(zhuān)業(yè)����、技術(shù)和測(cè)試專(zhuān)業(yè)知識(shí)以及能力。然而�����,應(yīng)注意�����,支持對(duì)違規(guī)企業(yè)或藥品采取監(jiān)管或行政行動(dòng)并不需要 API 的物理樣品�。
PART V–REGULATORY/ADMINISTRATIVE STRATEY
第五部分-監(jiān)管 / 行政策略
If one or more systems are documented as not in a state of control, OII should endorse an OAI inspection report.
如果一個(gè)或多個(gè)系統(tǒng)被記錄為未處于受控狀態(tài)�����,OII 應(yīng)認(rèn)可 OAI 檢查報(bào)告�����。
Normally, issuing a warning letter or taking other regulatory or administrative action should result in all profile classes being classified as unacceptable. However, if CDER does not approve a recommendation for a warning letter or other regulatory action, all profile classes should be classified as acceptable.
通常,發(fā)布警告信或采取其他監(jiān)管或行政行動(dòng)應(yīng)導(dǎo)致所有工藝類(lèi)別被歸類(lèi)為不可接受�。然而��,如果 CDER 不批準(zhǔn)關(guān)于警告信或其他監(jiān)管行動(dòng)的建議�����,則所有工藝類(lèi)別應(yīng)被歸類(lèi)為可接受�。
If an establishment with approved established conditions has an inspection that is classified as OAI and raises significant concerns about the quality system (particularly about the change management system), CDER offices will collaboratively evaluate the significant findings and the firm’s response. They will use this information to determine how these findings could potentially affect the approved established conditions.
如果具有已批準(zhǔn)既定條件的機(jī)構(gòu)的檢查被歸類(lèi)為 OAI���,且對(duì)質(zhì)量系統(tǒng)(特別是變更管理系統(tǒng))提出重大擔(dān)憂(yōu),CDER 各辦公室將合作評(píng)估重大發(fā)現(xiàn)和企業(yè)的回應(yīng)�����。他們將利用這些信息確定這些發(fā)現(xiàn)可能如何影響已批準(zhǔn)的既定條件�����。
Records, documents, and other information that are requested and reviewed during remote regulatory assessments may reveal potentially violative practices. In such cases, OMQ’s evaluation of a potential OAI recommendation will align with the procedures in this section during review of the case.
在遠(yuǎn)程監(jiān)管評(píng)估期間請(qǐng)求和審查的記錄�����、文件和其他信息可能揭示潛在的違規(guī)做法��。在這種情況下��,OMQ 對(duì)潛在 OAI 建議的評(píng)估將在審查案件期間遵循本節(jié)中的程序�����。
Recommendations for regulatory action for API CGMP deficiencies should cite the statute (section 501(a)(2)(B) of the FD&C Act) and not the drug product regulations in parts 210and 211. These recommendations should not cite ICH Q7; however, they can use ICH Q7as a guideline for describing the deficiencies. The regulatory action should demonstrate how the observed deficiencies could potentially impact or have already impacted the quality of the API. When evaluating whether to recommend regulatory or administrative action, consider the critical attributes of the API, the significance of its pharmacological activity, and the intended use of the drug product that will contain the API.
針對(duì) API CGMP 缺陷的監(jiān)管行動(dòng)建議應(yīng)引用法規(guī)(《FD&C 法案》第 501 (a)(2)(B) 條)����,而非第 210和211部分中的藥品法規(guī)。這些建議不應(yīng)引用 ICH Q7�;然而,它們可以使用 ICH Q7 作為描述缺陷的指南�。監(jiān)管行動(dòng)應(yīng)證明所觀察到的缺陷如何可能影響或已經(jīng)影響 API 的質(zhì)量。在評(píng)估是否建議采取監(jiān)管或行政行動(dòng)時(shí)���,應(yīng)考慮 API 的關(guān)鍵屬性、其藥理活性的重要性以及將包含該 API 的藥品的預(yù)期用途��。
Evidence that supports a significant deficiency or pattern of deficiencies within a system may indicate system failure. The failure of a system puts all of the manufacturer’s APIs at risk and should be promptly corrected. The following are examples of deficiencies that may result in an inspection that is classified as OAI:
證明系統(tǒng)內(nèi)存在重大缺陷或缺陷模式的證據(jù)可能表明系統(tǒng)故障�����。系統(tǒng)故障會(huì)使制造商的所有API 面臨風(fēng)險(xiǎn)����,應(yīng)立即糾正。以下是可能導(dǎo)致檢查被歸類(lèi)為 OAI 的缺陷示例:
1.Contamination of APIs with filth, objectionable microorganisms, toxic chemicals, or significant amounts of other types of chemicals, or a reasonable potential for such contamination because of a demonstrated route of contamination (facilities and equipment system; production system).
API 受到污物����、不良微生物、有毒化學(xué)品或大量其他類(lèi)型化學(xué)品的污染����,或由于已證明的污染途徑而存在此類(lèi)污染的合理可能性(設(shè)施和設(shè)備系統(tǒng)�;生產(chǎn)系統(tǒng))�。
2.Failure to show that API batches conform to established specifications, such as specifications in drug applications, USP specifications, customer specifications, or label claims (quality system).未能證明 API 批次符合既定規(guī)格,如藥品申請(qǐng)中的規(guī)格����、USP 規(guī)格、客戶(hù)規(guī)格或標(biāo)簽聲明(質(zhì)量系統(tǒng))�。
3.Failure to ensure the accuracy and integrity of data. Complete, consistent, and accurate data should be attributable, legible, contemporaneously recorded, original or a true copy, and accurate. Examples of data integrity concerns include failing to scientifically justify not reporting relevant data, altering of raw data, inconsistently documenting manufacturing operations, backdating test results, testing into compliance, and fabricating test results. Examples of failure to ensure the accuracy and integrity of data include systems which allow for the alteration or deletion of raw data, systems without audit trails activated, loose raw data forms without adequate issuance and reconciliation, and inadequate risk-based monitoring for data integrity concerns (all systems).
未能確保數(shù)據(jù)的準(zhǔn)確性和完整性。完整���、一致和準(zhǔn)確的數(shù)據(jù)應(yīng)可歸因����、清晰可讀����、同期記錄、原始或真實(shí)副本��,并且準(zhǔn)確����。數(shù)據(jù)完整性問(wèn)題的示例包括未能科學(xué)證明不報(bào)告相關(guān)數(shù)據(jù)的合理性、更改原始數(shù)據(jù)、不一致地記錄生產(chǎn)操作����、追溯測(cè)試結(jié)果�、測(cè)試至合格以及偽造測(cè)試結(jié)果。未能確保數(shù)據(jù)準(zhǔn)確性和完整性的示例包括允許更改或刪除原始數(shù)據(jù)的系統(tǒng)���、未激活審計(jì)追蹤的系統(tǒng)��、沒(méi)有充分發(fā)放和核對(duì)的松散原始數(shù)據(jù)表格�����,以及對(duì)數(shù)據(jù)完整性問(wèn)題的基于風(fēng)險(xiǎn)的監(jiān)控不足(所有系統(tǒng))�。
4.Failure to comply with commitments in drug applications or DMFs. All of the required information in these commitments should be accurate and current. This includes information about the manufacturing process, impurity profiles, and other specifications or proceduresassociated with the manufacture of the API (quality system).
未能遵守藥品申請(qǐng)或 DMF 中的承諾�����。這些承諾中的所有所需信息都應(yīng)準(zhǔn)確且最新���。這包括與 API 生產(chǎn)相關(guān)的生產(chǎn)過(guò)程����、雜質(zhì)概況以及其他規(guī)格或程序的信息(質(zhì)量系統(tǒng))。
5.Distribution of an API that does not conform to established specifications (quality system).
分銷(xiāo)不符合既定規(guī)格的 API(質(zhì)量系統(tǒng))����。
6.Deliberate blending of API batches in an attempt to: (1) dilute or hide filth or other noxious contaminants; or(2) disguise a critical quality defect and obtain a batch that meets its specifications (production system).
故意混合 API 批次,試圖:(1)稀釋或隱藏污物或其他有害污染物����;或(2)掩蓋關(guān)鍵質(zhì)量缺陷并獲得符合其規(guī)格的批次(生產(chǎn)系統(tǒng))。
7.Failure to demonstrate that all materials (including water and any other solvents used in the final step of the API production process) are chemically and microbiologically suitable for their intended use and do not adversely affect the quality of the API (materials system).
未能證明所有物料(包括在 API 生產(chǎn)過(guò)程最后一步中使用的水和任何其他溶劑)在化學(xué)和微生物方面適合其預(yù)期用途�,且不會(huì)對(duì) API 質(zhì)量產(chǎn)生不利影響(物料系統(tǒng))。
8. Lack of adequate validation for critical steps in the API production process, particularly for the final separation and purification of the API or API production processes for whichthere is evidence that the process is not adequately controlled. Lack of adequate control may be indicated by repeated batch failures or wide variation in final yields compared with the process average over time (quality system; production system).
API 生產(chǎn)過(guò)程中關(guān)鍵步驟缺乏充分的驗(yàn)證���,特別是 API 的最終分離和純化或有證據(jù)表明過(guò)程未得到充分控制的 API 生產(chǎn)過(guò)程����??刂撇蛔憧赡鼙憩F(xiàn)為重復(fù)的批次失敗或與過(guò)程平均值相比最終收率的廣泛變化(質(zhì)量系統(tǒng);生產(chǎn)系統(tǒng))��。
9.Implementation of retrospective process validation for an existing API production processwhen the process has changed significantly, the firm lacks impurity profile data, or there is evidence of repeated batch failures because of process variability (quality system; production system).
當(dāng)現(xiàn)有 API 生產(chǎn)過(guò)程發(fā)生重大變化����、企業(yè)缺乏雜質(zhì)概況數(shù)據(jù)或有證據(jù)表明由于過(guò)程變異性導(dǎo)致重復(fù)批次失敗時(shí),對(duì)現(xiàn)有 API 生產(chǎn)過(guò)程實(shí)施回顧性過(guò)程驗(yàn)證(質(zhì)量系統(tǒng)����;生產(chǎn)系統(tǒng))����。
10.Failure to establish an impurity profile for each API production process. FDA expects manufacturers to establish complete impurity profiles for each API as part of the process validation effort. This includes collecting data on: (1) actual and potential organic impurities that may arise during synthesis, purification, and storage of the API; (2) actual and potential inorganic impurities that may arise during the API production process; and (3) organic and inorganic solvents used during the manufacturing process that are known to carry over to the API. Impurity profile testing of each batch or after a specified number of batches may detect new impurities that could be from a deliberate or nondeliberate change inthe API manufacturing process (laboratory control system).
未能為每個(gè) API 生產(chǎn)過(guò)程建立雜質(zhì)概況�。FDA 期望制造商作為過(guò)程驗(yàn)證工作的一部分����,為每個(gè) API建立完整的雜質(zhì)概況。這包括收集以下數(shù)據(jù):(1)在 API 合成�����、純化和儲(chǔ)存期間可能產(chǎn)生的實(shí)際和潛在有機(jī)雜質(zhì)����;(2)在 API 生產(chǎn)過(guò)程中可能產(chǎn)生的實(shí)際和潛在無(wú)機(jī)雜質(zhì);(3)在生產(chǎn)過(guò)程中使用的����、已知會(huì)帶入 API 中的有機(jī)和無(wú)機(jī)溶劑。每批或特定數(shù)量批次后的雜質(zhì)概況測(cè)試可能檢測(cè)到新的雜質(zhì)�����,這些雜質(zhì)可能來(lái)自 API 生產(chǎn)過(guò)程中的故意或非故意變更(實(shí)驗(yàn)室控制系統(tǒng))。
11.Failure to show that a reprocessed batch complies with all of the established standards, specifications, and characteristics (quality system; laboratory control system).
未能證明返工批次符合所有既定標(biāo)準(zhǔn)����、規(guī)格和特性(質(zhì)量系統(tǒng);實(shí)驗(yàn)室控制系統(tǒng))��。
12.Failure to test for residues of organic or inorganic solvents used during manufacturing that may carry over to the API using analytical procedures with appropriate levels of sensitivity (laboratory control system).
未能使用具有適當(dāng)靈敏度水平的分析程序測(cè)試生產(chǎn)過(guò)程中使用的可能帶入 API 中的有機(jī)或無(wú)機(jī)溶劑殘留(實(shí)驗(yàn)室控制系統(tǒng))�。
13.Failure to have a formal process change control system in place to evaluate changes in starting materials, facilities, support systems, equipment, processing steps, and packaging materials that may affect the quality of APIs (all systems).
缺乏正式的過(guò)程變更控制系統(tǒng)來(lái)評(píng)估起始物料、設(shè)施����、支持系統(tǒng)、設(shè)備��、加工步驟和包裝材料的變更可能對(duì) API 質(zhì)量產(chǎn)生的影響(所有系統(tǒng))����。
14.Failure to maintain batch and quality control records (quality system).
未能維護(hù)批次和質(zhì)量控制記錄(質(zhì)量系統(tǒng))。
15.Incomplete stability studies to establish API stability for the intended period of use, forexample, failure to conduct forced degradation studies on APIs to isolate, identify, and quantify potential degradants that may arise during storage (laboratory control system).
用于確定 API 在預(yù)期使用期限內(nèi)穩(wěn)定性的穩(wěn)定性研究不完整����,例如,未能對(duì) API 進(jìn)行強(qiáng)制降解研究以分離�����、識(shí)別和量化儲(chǔ)存期間可能產(chǎn)生的潛在降解產(chǎn)物(實(shí)驗(yàn)室控制系統(tǒng))。
16.Use of laboratory test methods that are inadequate or have not been validated, or the use of an inadequately qualified or untraceable reference standard (laboratory control system).
使用不充分或未經(jīng)驗(yàn)證的實(shí)驗(yàn)室測(cè)試方法�,或使用資質(zhì)不足或不可追溯的參考標(biāo)準(zhǔn)品(實(shí)驗(yàn)室控制系統(tǒng))。
17.Packaging and labeling in a way that introduces a significant risk of mislabeling (packaging and labeling system).
以可能導(dǎo)致錯(cuò)誤貼標(biāo)簽的方式進(jìn)行包裝和貼標(biāo)簽(包裝和貼標(biāo)簽系統(tǒng))�����。