為藥品上市許可持有人和/或生產(chǎn)企業(yè)對(duì)已上市中藥進(jìn)行藥學(xué)變更研究提供可參考的技術(shù)標(biāo)準(zhǔn)�,國(guó)家藥品監(jiān)督管理局藥品審評(píng)中心于2021年4月發(fā)布了《已上市中藥藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》����。其核心是基于風(fēng)險(xiǎn)控制理念�,確保藥品安全�、有效、質(zhì)量可控����,指導(dǎo)針對(duì)生產(chǎn)、質(zhì)量控制���、使用等環(huán)節(jié)擬進(jìn)行的變更開展研究和評(píng)估工作���。本文就基于此指導(dǎo)原則進(jìn)行總結(jié)剖析,以期為讀者提供一定的幫助和指導(dǎo)����。
01概述
● 適用范圍: 涵蓋七大類主要變更事項(xiàng):變更生產(chǎn)工藝、變更制劑處方中的輔料����、變更規(guī)格或包裝規(guī)格、變更注冊(cè)標(biāo)準(zhǔn)�、變更包裝材料和容器、變更有效期或貯藏條件、變更制劑生產(chǎn)場(chǎng)地����。其他變更需參照基本原則執(zhí)行����。
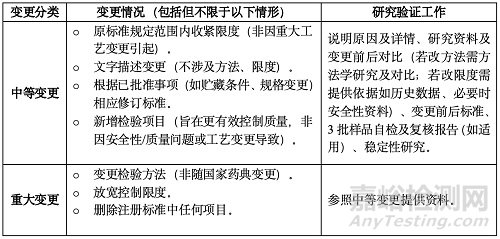
● 變更分類(基于風(fēng)險(xiǎn)與影響程度):
o 重大變更: 可能對(duì)藥品安全性、有效性和質(zhì)量可控性產(chǎn)生重大影響的變更����。
o 中等變更: 可能對(duì)藥品安全性、有效性和質(zhì)量可控性產(chǎn)生中等程度影響的變更����。
o 微小變更: 對(duì)藥品安全性、有效性和質(zhì)量可控性基本不產(chǎn)生影響的變更�。分類不明確時(shí),持有人需根據(jù)藥品特點(diǎn)和研究評(píng)估結(jié)果確定類別���。
● 制定基礎(chǔ)與理念: 以國(guó)家法規(guī)為指導(dǎo)����,總結(jié)中藥變更研究經(jīng)驗(yàn)���,結(jié)合中藥特點(diǎn)列舉常見變更及分類�。鼓勵(lì)借鑒ICH的“質(zhì)量源于設(shè)計(jì)”、“設(shè)計(jì)空間”����、“既定條件”等理念。
強(qiáng)調(diào)持有人是變更研究及評(píng)估的責(zé)任主體����,應(yīng)基于科學(xué)評(píng)估決定變更實(shí)施。特殊變更或新技術(shù)應(yīng)用問題�,提倡與監(jiān)管機(jī)構(gòu)溝通。
02基本原則
1.持有人應(yīng)履行主體責(zé)任: 持有人需全面了解藥品����,建立全生命周期質(zhì)量風(fēng)險(xiǎn)管理體系。變更時(shí)須明確原因����、程度及影響,依規(guī)開展研究���,全面分析結(jié)果����,評(píng)估對(duì)安全、有效���、質(zhì)量可控性的影響����,并按規(guī)定提出補(bǔ)充申請(qǐng)���、備案或報(bào)告。
2.變更應(yīng)必要����、科學(xué)、合理: 變更應(yīng)基于藥品知識(shí)積累更新(如生產(chǎn)經(jīng)驗(yàn)���、質(zhì)量回顧���、新技術(shù)應(yīng)用等),以科學(xué)方法和決策提升生產(chǎn)質(zhì)量���、便利患者為目的����。研究應(yīng)以既往研究和生產(chǎn)數(shù)據(jù)為基礎(chǔ),前期研究數(shù)據(jù)可為后期變更提供依據(jù)����。研究越系統(tǒng)深入,數(shù)據(jù)越充分���,對(duì)變更評(píng)估越有利�。需說明變更的必要性����、科學(xué)性和合理性。
3.全面評(píng)估���、驗(yàn)證變更影響: 中藥質(zhì)量依賴全過程控制�,各環(huán)節(jié)緊密關(guān)聯(lián)�,任一變更(處方、工藝���、場(chǎng)地����、標(biāo)準(zhǔn)等)可能產(chǎn)生全面影響�。
o 需全面研究評(píng)估變更對(duì)安全���、有效、質(zhì)量可控性的風(fēng)險(xiǎn)與影響程度�。
o 根據(jù)變更具體情況、藥物性質(zhì)及制劑要求選擇針對(duì)性指標(biāo)評(píng)估影響����。
o 變更后藥品應(yīng)質(zhì)量可控、均一穩(wěn)定����。
o 變更不得引起藥用物質(zhì)基礎(chǔ)或制劑吸收�、利用的明顯改變,不得對(duì)安全有效性產(chǎn)生不利影響或帶來(lái)明顯變化����;否則需進(jìn)行全面安全有效性評(píng)價(jià)。若工藝或輔料變更導(dǎo)致物質(zhì)基礎(chǔ)或吸收利用明顯改變�,應(yīng)按改良型新藥研究。
4.遵循中醫(yī)藥自身特點(diǎn)和規(guī)律: 中藥變更應(yīng)基于中醫(yī)藥理論和傳統(tǒng)工藝����,在工藝方法不變的前提下,工藝參數(shù)變更一般可通過藥學(xué)研究(如比較出膏率/干膏率�、浸出物�、指紋圖譜/特征圖譜����、多成份含量)評(píng)估變更前后一致性。
03基本要求
1.研究用樣品要求: 研究樣品應(yīng)能代表生產(chǎn)實(shí)際情況����。工藝驗(yàn)證需用生產(chǎn)規(guī)模樣品。變更前后質(zhì)量對(duì)比研究通常采用變更前和變更后各連續(xù)3批樣品���。
2.關(guān)聯(lián)變更要求: 一項(xiàng)變更可能伴隨其他變更(如規(guī)格變更可能伴隨輔料或包材變更)�,稱為關(guān)聯(lián)變更�。研究工作需綜合考慮各項(xiàng)變更的基本思路,按技術(shù)要求最高的變更類別進(jìn)行�。
3.含毒性藥味制劑要求: 此類制劑變更須特別關(guān)注安全性,尤其針對(duì):
o 含大毒(劇毒)藥味的制劑(依據(jù)國(guó)務(wù)院《醫(yī)療用毒性藥品管理辦法》28種及各級(jí)標(biāo)準(zhǔn)標(biāo)注���,毒性認(rèn)定以高標(biāo)準(zhǔn)為準(zhǔn))�。
o 含現(xiàn)代研究發(fā)現(xiàn)有嚴(yán)重毒性藥味的制劑����。
o 含分類為有毒藥味,且用于兒科����、妊娠期和哺乳期婦女的制劑���。
o 含孕婦禁用/慎用藥味,且功能主治為妊娠期和哺乳期婦女的制劑�。
4.質(zhì)量對(duì)比研究要求: 是變更研究考量及分類的重要依據(jù)。若現(xiàn)行標(biāo)準(zhǔn)難以充分反映質(zhì)量或可控性低�,應(yīng)開展質(zhì)量及標(biāo)準(zhǔn)研究,采用合適指標(biāo)(如浸出物�、指紋圖譜/特征圖譜、溶出度�、生物活性測(cè)定等)進(jìn)行對(duì)比研究,客觀評(píng)估變更影響����。
5.其他: 中西復(fù)方制劑�、中藥注射劑、緩釋/控釋制劑等的變更����,應(yīng)充分考慮其特點(diǎn)和制劑要求,全面評(píng)估影響����,參照相關(guān)技術(shù)指導(dǎo)原則進(jìn)行�。
04各類變更具體要求
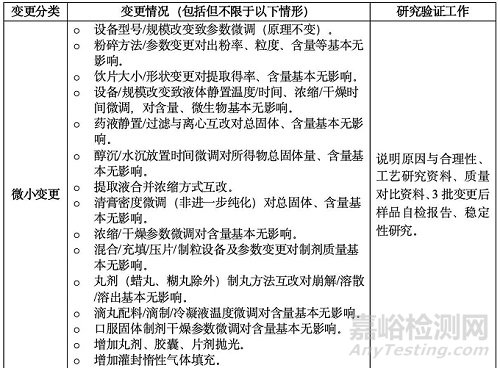
(一)變更生產(chǎn)工藝
涉及工藝路線����、方法、參數(shù)等變更�,可能出現(xiàn)在前處理、提取�、分離純化、濃縮����、干燥、制劑成型等環(huán)節(jié)����。設(shè)備變更需評(píng)估其對(duì)工藝和質(zhì)量的影響。核心原則:工藝變更不應(yīng)引起藥用物質(zhì)基礎(chǔ)的明顯改變�,否則需全面評(píng)價(jià)。
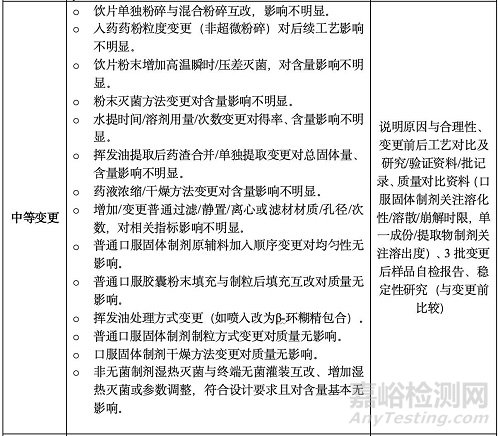
(二)變更制劑處方中的輔料
包括變更供應(yīng)商���、種類�、用量或級(jí)別����。重點(diǎn)關(guān)注輔料性質(zhì)(是否影響溶出/釋放或?yàn)橛绊懼苿w內(nèi)藥物吸收的關(guān)鍵輔料)和制劑特性���。關(guān)聯(lián)變更按高要求類別研究。使用新輔料需按新輔料要求研究���。明顯影響吸收利用的輔料變更需全面評(píng)價(jià)(如特殊輔料蜂蜜/冰糖改變且其功能主治與藥品相關(guān)�;外用制劑增刪顯著影響吸收����、利用的輔料)。微小和中等變更涉及的輔料應(yīng)為常用輔料�,有國(guó)標(biāo)或注冊(cè)標(biāo)準(zhǔn),登記狀態(tài)需為“A”����。
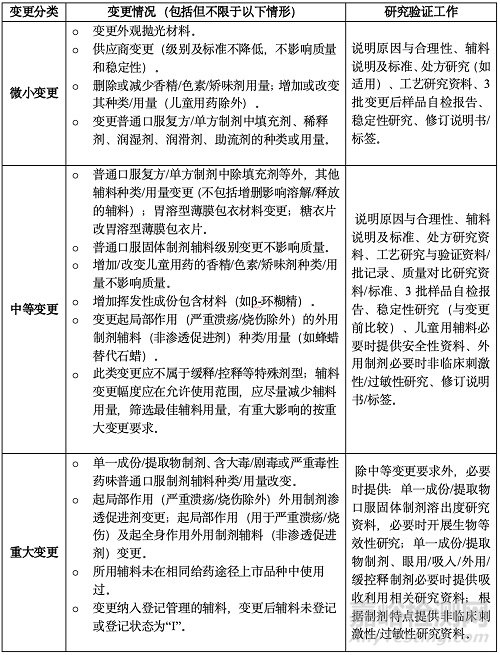
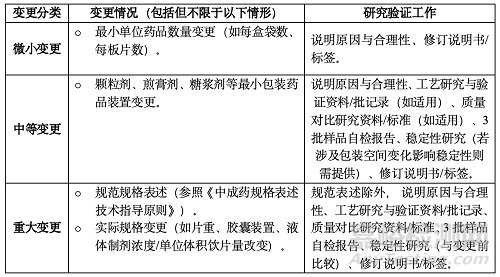
(三)變更規(guī)格或包裝規(guī)格
變更規(guī)格應(yīng)科學(xué)、合理����、必要����、方便臨床用藥,符合用法用量���。變更不得引起藥用物質(zhì)基礎(chǔ)變化����,不得改變?cè)梅ㄓ昧炕蜻m用人群。否則需全面評(píng)價(jià)����。
(四)變更注冊(cè)標(biāo)準(zhǔn)
主要指注冊(cè)標(biāo)準(zhǔn)中檢驗(yàn)項(xiàng)目、方法或限度/范圍的修訂����。修改后標(biāo)準(zhǔn)應(yīng)不低于國(guó)家藥品標(biāo)準(zhǔn)。變更不應(yīng)降低質(zhì)量控制水平����。工作重點(diǎn)是方法學(xué)研究和驗(yàn)證、限度確定���。需關(guān)注變更是否影響有效期(如提高標(biāo)準(zhǔn)需考察原有效期是否符合新標(biāo)準(zhǔn))���。
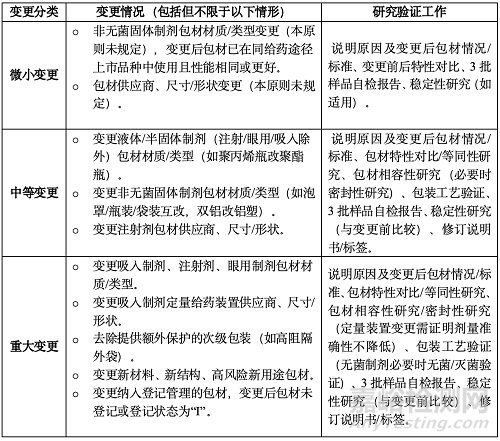
(五)變更包裝材料和容器
主要指直接接觸藥品的包裝。風(fēng)險(xiǎn)取決于給藥途徑�、包材性能、包材-制劑相容性。變更應(yīng)有益于保證質(zhì)量和穩(wěn)定性�,至少不降低保護(hù)作用,不得發(fā)生不良相互作用����。重點(diǎn)研究相互作用和穩(wěn)定性影響。中間體包材變更按品種要求評(píng)估�。
(六)變更有效期或貯藏條件
包含延長(zhǎng)/縮短有效期、嚴(yán)格/放寬貯藏條件�。需注意可能涉及多種情況。若穩(wěn)定性方案�、質(zhì)控項(xiàng)目/方法、工藝/處方等變更����,需相應(yīng)研究。擬延長(zhǎng)的有效期不得超過長(zhǎng)期穩(wěn)定性考察時(shí)間�。關(guān)注中間體貯藏時(shí)間/條件變更。
(七)變更制劑生產(chǎn)場(chǎng)地
指制劑實(shí)際生產(chǎn)地址改變或新增�,或同一地址內(nèi)場(chǎng)地改建/重建/新建(需標(biāo)明同一物理地址)。包括持有人自有或受托方場(chǎng)地����。提取物場(chǎng)地變更要求同制劑。
·核心要求: 確保變更后持續(xù)穩(wěn)定生產(chǎn)符合要求的藥品�。不得改變處方、工藝�、直接接觸包材,不得降低過程控制水平及標(biāo)準(zhǔn)�。需全面驗(yàn)證,重點(diǎn)考察變更前后全過程質(zhì)量控制一致性���,通過關(guān)鍵工藝參數(shù)�、藥用物質(zhì)基礎(chǔ)對(duì)比判定質(zhì)量差異�。
·法規(guī)依據(jù): 執(zhí)行《藥品生產(chǎn)監(jiān)督管理辦法》《藥品上市后變更管理辦法(試行)》。
·研究驗(yàn)證: 變更情況及原因�、新舊場(chǎng)地工藝比較(設(shè)備性能、原理����、能力、廠家���、型號(hào))�、質(zhì)量風(fēng)險(xiǎn)評(píng)估���、工藝研究與驗(yàn)證資料/批記錄(如適用)�、質(zhì)量對(duì)比研究(如適用)����、3批樣品自檢報(bào)告(如適用)�、穩(wěn)定性研究(與變更前比較���,如適用)����。
05總結(jié)
該指導(dǎo)原則構(gòu)建了以風(fēng)險(xiǎn)控制為核心���、基于變更對(duì)藥品安全�、有效�、質(zhì)量可控性影響程度的三級(jí)分類管理體系(重大、中等�、微小)�。它明確了持有人作為變更責(zé)任主體的要求,強(qiáng)調(diào)了變更的必要性����、科學(xué)性和合理性原則,以及遵循中醫(yī)藥特點(diǎn)的重要性����。通過詳細(xì)規(guī)定七大類主要變更事項(xiàng)的具體情形����、研究驗(yàn)證要求及注意事項(xiàng)���,為中藥上市后藥學(xué)變更的系統(tǒng)化、規(guī)范化管理提供了技術(shù)依據(jù)����,旨在保障變更后藥品質(zhì)量不降低、風(fēng)險(xiǎn)可控�,最終維護(hù)公眾用藥安全有效。
參考文獻(xiàn)
[1] CDE����,已上市中藥藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行),2021年4月