一 范圍
本文僅適用于化學(xué)藥品Ⅲ期臨床試驗(yàn)期間及上市后增加規(guī)格的補(bǔ)充申請(qǐng),不適用于一致性評(píng)價(jià)品種改規(guī)格等情況�。
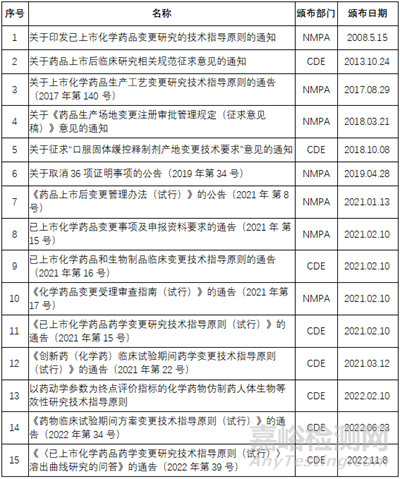
二 法規(guī)要求
三 增加規(guī)格的變更分類
根據(jù)《創(chuàng)新藥(化學(xué)藥)臨床試驗(yàn)期間藥學(xué)變更技術(shù)指導(dǎo)原則(試行)》���、《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》、《已上市化學(xué)藥品和生物制品臨床變更技術(shù)指導(dǎo)原則》等的分類�,化學(xué)藥品增加規(guī)格分為幾種情況���,如下圖:
四 增加規(guī)格的研究驗(yàn)證工作
4.1 Ⅲ期臨床期間增加規(guī)格
臨床試驗(yàn)期間規(guī)格變更可能對(duì)產(chǎn)品安全性和質(zhì)量產(chǎn)生較顯著的影響,通常應(yīng)視為重大變更���。規(guī)格發(fā)生變更時(shí),通常伴隨著處方���、工藝的變更�?���?傮w上規(guī)格變更研究思路與處方工藝變更類似。此外�,規(guī)格變更和劑型也是密切相關(guān)。
參考《已上市化學(xué)藥品生產(chǎn)工藝變更研究技術(shù)指導(dǎo)原則》和《已上市化學(xué)藥品變更研究技術(shù)指導(dǎo)原則(試行)》等����。
比如基于Ⅱ期臨床試驗(yàn)結(jié)果確定了給藥劑量�,在Ⅲ期臨床階段擬增加新的規(guī)格,需要根據(jù)產(chǎn)品的特性���,通過體外研究(藥學(xué)相關(guān)對(duì)比等)和(或)體內(nèi)試驗(yàn)(如BE�、PK 等)來評(píng)估新規(guī)格同原規(guī)格之間的可銜接性����。
通常���,在創(chuàng)新藥研究進(jìn)程中�,越是研究后期發(fā)生的變更,越需要開展細(xì)致深入的研究�,以證明變更的可接受性����。創(chuàng)新藥在完成支持上市的關(guān)鍵臨床研究后�,如發(fā)生重大的藥學(xué)變更,需慎重考慮���。
4.2 已批準(zhǔn)用法用量范圍內(nèi)增加規(guī)格
增加規(guī)格應(yīng)遵循方便臨床用藥的原則,應(yīng)有合理����、科學(xué)的依據(jù)?���?傮w上���,增加規(guī)格一般應(yīng)在其臨床使用的用法用量范圍內(nèi)����,不得大于單次用藥的最高劑量���,或?qū)Τ扇擞盟巵碚f不得小于成人單次用藥的最低劑量�。
《已上市化學(xué)藥品藥學(xué)變更研究技術(shù)指導(dǎo)原則(試行)》涵蓋的增加規(guī)格����,是指片劑/膠囊劑等單劑量藥品的主藥在單劑量處方中的標(biāo)示量�、注射劑的單劑量包裝中主藥標(biāo)示量/濃度、非無菌半固體制劑/口服溶液劑/滴眼劑等制劑的處方中藥物濃度的改變���。新增規(guī)格應(yīng)為原研藥品增加的新規(guī)格或仿制藥增加目前原研藥品/參比制劑已有的規(guī)格,同時(shí)不得改變藥品原批準(zhǔn)的適應(yīng)癥�、用法用量或者適用人群等。增加規(guī)格均屬于藥學(xué)重大變更����。
對(duì)于非無菌半固體制劑/口服液體劑/滴眼劑等制劑的多劑量包裝變更為單劑量包裝也按增加規(guī)格申報(bào)�。
4.2.1 研究驗(yàn)證工作
1����、說明變更的具體情況和原因。
2、對(duì)新增規(guī)格進(jìn)行處方研究���、工藝研究和/或驗(yàn)證�,并與變更前規(guī)格進(jìn)行對(duì)比�。對(duì)于無菌制劑���,還需進(jìn)行無菌/滅菌工藝驗(yàn)證����。
3�、提供變更后一批樣品的批生產(chǎn)記錄���。
4、對(duì)變更規(guī)格前后的樣品進(jìn)行質(zhì)量對(duì)比研究���,重點(diǎn)比較變更前后樣品的溶出曲線���、雜質(zhì)譜、關(guān)鍵理化性質(zhì)等 ,應(yīng)符合相關(guān)指導(dǎo)原則的要求���。
5�、對(duì)變更后連續(xù)生產(chǎn)的三批樣品進(jìn)行檢驗(yàn) 應(yīng)符合質(zhì)量標(biāo)準(zhǔn)的規(guī)定 。
6、對(duì)變更后三批樣品進(jìn)行加速及長期穩(wěn)定性考察����,申請(qǐng)時(shí)提供3 ~ 6 個(gè)月的穩(wěn)定性研究資料����,并與原規(guī)格產(chǎn)品的穩(wěn)定性情況進(jìn)行比較�。
7���、應(yīng)參考《以藥動(dòng)學(xué)參數(shù)為終點(diǎn)評(píng)價(jià)指標(biāo)的化學(xué)藥物仿制藥人體生物等效性研究技術(shù)指導(dǎo)原則》及其他相關(guān)指導(dǎo)原則,考慮新增規(guī)格是否需進(jìn)行生物等效性研究�。
4.2.2 案例分享
a.劑型問題
劑型合理性應(yīng)在明確藥物理化性質(zhì)及生物學(xué)性質(zhì)的基礎(chǔ)上,結(jié)合藥物臨床治療需求進(jìn)行分析�。目前對(duì)注射劑劑型合理性和滅菌工藝的技術(shù)要求����,在遵循劑型選擇一般原則的基礎(chǔ)上���,從無菌保證水平考慮���,注射劑要首選可采用終端滅菌工藝的劑型����。若目前已有采用終端滅菌工藝的同品種注射劑上市,對(duì)采用無菌生產(chǎn)工藝的凍干粉針劑�,其無菌保證水平低于已上市產(chǎn)品,一般不再批準(zhǔn)其補(bǔ)充申請(qǐng)���。同時(shí)藥品劑型為不合理劑型時(shí)���,其增加規(guī)格的補(bǔ)充申請(qǐng)將不予認(rèn)可。
b.處方���、工藝研究
劑型�、規(guī)格發(fā)生變更時(shí)�,通常伴隨著處方�、工藝的變更���。如增加薄膜衣規(guī)格�,既新增薄膜包衣工藝又是關(guān)鍵生產(chǎn)工藝���,研究者應(yīng)對(duì)包衣工藝進(jìn)行研究和驗(yàn)證����,并確定關(guān)鍵工藝影響因素和工藝參數(shù)����;如注射劑新增規(guī)格會(huì)對(duì)滅菌工藝帶來影響,會(huì)影響滅菌過程藥液中的熱分布�,應(yīng)對(duì)新增規(guī)格產(chǎn)品進(jìn)行滅菌工藝驗(yàn)證。
4.3 未在已批準(zhǔn)用法用量范圍內(nèi)增加規(guī)格
對(duì)于未在已批準(zhǔn)用法用量范圍內(nèi)增加新規(guī)格的藥品����,增加新規(guī)格通常在用法用量變更(給藥劑量超過或低于已批準(zhǔn)的用法用量范圍)的同時(shí)提出�,此類變更涉及藥品安全和有效信息的變更����,屬于臨床重大A類變更����,是指與用藥人群�、有效性���、安全性����、給藥劑量和給藥方法相關(guān)的變更�。
《已上市化學(xué)藥品和生物制品臨床變更技術(shù)指導(dǎo)原則》指出臨床重大A類變更對(duì)藥品安全有效使用產(chǎn)生的影響及風(fēng)險(xiǎn)程度較高���,通常需要嚴(yán)格設(shè)計(jì)并實(shí)施的臨床試驗(yàn)數(shù)據(jù)和/或非臨床研究數(shù)據(jù)支持�。藥品上市許可持有人在開展臨床試驗(yàn)前,應(yīng)首先提出補(bǔ)充申請(qǐng)�,在獲得批準(zhǔn)后方可開展臨床試驗(yàn)����。藥品上市許可持有人完成臨床試驗(yàn)并經(jīng)評(píng)估認(rèn)為試驗(yàn)數(shù)據(jù)可支持相應(yīng)變更時(shí),可向國家藥品監(jiān)督管理局遞交補(bǔ)充申請(qǐng)����。
4.3.1 藥學(xué)研究技術(shù)要求
參考4.2.1的1-6。
4.3.2 臨床研究技術(shù)要求
臨床變更是否需要開展研究以及研究的復(fù)雜程度取決于變更對(duì)藥品安全性�、有效性及臨床安全有效使用的潛在影響應(yīng)具體問題具體分析。就支持重大變更所需的臨床和/或非臨床數(shù)據(jù)的充分性鼓勵(lì)藥品上市許可持有人與國家藥品監(jiān)督管理局進(jìn)行溝通交流�。
通常�,對(duì)于重大變更A類 ����,所需支持性非臨床和/或臨床安全性和有效性研究的類型和范圍應(yīng)基于變更相關(guān)的獲益/風(fēng)險(xiǎn)評(píng)估、藥品特征�、已批準(zhǔn)適應(yīng)癥特點(diǎn)(發(fā)病率、死亡率���、急性或慢性疾病���、當(dāng)前疾病治療的可及性等)����、安全性因素等方面綜合評(píng)估后確定����。例如:對(duì)于用法用量的變更,應(yīng)在評(píng)估已有劑量相關(guān)安全有效性數(shù)據(jù)并確保受試者安全的基礎(chǔ)上���, 開展相應(yīng)研究 ,如變更前后不同用法用量下的安全有效性對(duì)比研究。
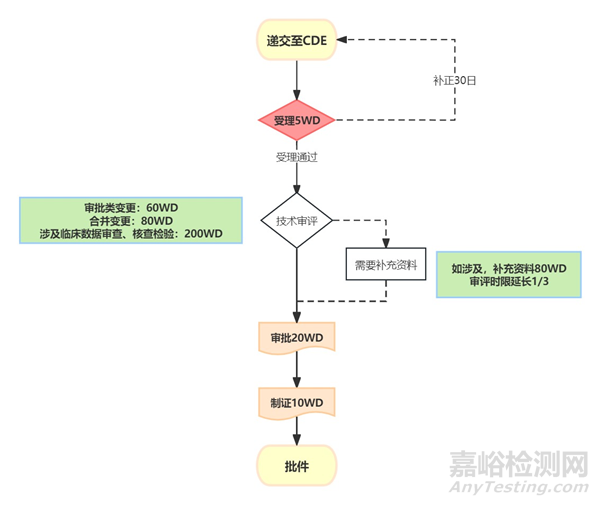
五 增加規(guī)格的補(bǔ)充申請(qǐng)流程及時(shí)限
增加規(guī)格的變更通常為重大變更�,按照《藥品注冊(cè)管理辦法》、《藥品上市后變更管理辦法(試行)》及相關(guān)指南和指導(dǎo)原則的要求���,增加規(guī)格的變更需要遞交國家藥品審評(píng)中心補(bǔ)充申請(qǐng),批準(zhǔn)后實(shí)施�。
重大變更*補(bǔ)充申請(qǐng)流程及時(shí)限如下圖:
*重大變更 A類:藥品上市許可持有人在開展臨床試驗(yàn)前,應(yīng)首先提出補(bǔ)充申請(qǐng)����,在獲得批準(zhǔn)后方可開展臨床試驗(yàn)。藥品上市許可持有人完成臨床試驗(yàn)并經(jīng)評(píng)估認(rèn)為試驗(yàn)數(shù)據(jù)可支持相應(yīng)變更時(shí)����,可向國家藥品監(jiān)督管理局遞交補(bǔ)充申請(qǐng)����。
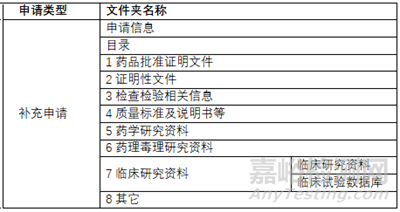
六 增加規(guī)格的補(bǔ)充申請(qǐng)申報(bào)資料要求
6.1 重大變更補(bǔ)充申請(qǐng)申報(bào)資料要求
根據(jù)關(guān)于發(fā)布已上市化學(xué)藥品變更事項(xiàng)及申報(bào)資料要求的通告(2021年 第15號(hào))的要求,藥品補(bǔ)充申請(qǐng)時(shí)應(yīng)按以下申報(bào)資料準(zhǔn)備補(bǔ)充申請(qǐng)����。
6.2 重大變更A類補(bǔ)充申請(qǐng)申報(bào)資料要求
根據(jù)已上市化學(xué)藥品變更事項(xiàng)及申報(bào)資料要求的通告(2021年 第15號(hào))和已上市化學(xué)藥品和生物制品臨床變更技術(shù)指導(dǎo)原則的要求���,重大變更A類補(bǔ)充申請(qǐng)申報(bào)資料要求如下:
在6.1 的基礎(chǔ)上,同時(shí)應(yīng)包含以下內(nèi)容:
1�、變更內(nèi)容及變更理由。
2、變更相關(guān)研究總結(jié)���。應(yīng)提供用于評(píng)估變更對(duì)藥品有效性和 /或安全性影響的主要研究方法及完成的研究情況���。
3����、與變更相關(guān)的非臨床研究資料和必要的國內(nèi)外文獻(xiàn)資料����。
4、變更相關(guān)的藥品臨床試驗(yàn)資料���,包括綜述資料����,臨床試驗(yàn)計(jì)劃和方案、統(tǒng)計(jì)分析計(jì)劃、臨床研究報(bào)告等���;臨床研究報(bào)告中應(yīng)對(duì)臨床測定方法和驗(yàn)證情況進(jìn)行詳細(xì)說明����。
5、申請(qǐng)變更的說明書和包裝標(biāo)簽樣稿���, 藥品 批件的說明書和包裝標(biāo)簽 附件 �、修訂說明、修訂前后對(duì)比表�。
6�、藥物警戒計(jì)劃,針對(duì)變更內(nèi)容及研究數(shù)據(jù)確定是否需要制定并提供相關(guān)藥物警戒計(jì)劃�。
上述2、3���、4相關(guān)資料可按照《M4:人用藥物注冊(cè)申請(qǐng)通用技術(shù)文檔(CTD)》中相關(guān)模塊要求撰寫����。
參考文獻(xiàn)
[1]. 馮欣;王建嬌;田曉娟;佟利家.變更藥品規(guī)格補(bǔ)充申請(qǐng)常見問題分析及建議[J].首都醫(yī)藥,2012,19(22):7.
[2]. 張豹子;田潔;寧黎麗.補(bǔ)充申請(qǐng)?jiān)黾右?guī)格常見問題分析[J].中國新藥雜志,2013,22(01):20-22.