前言
如果您還沒有建立 HPLC 方法,要進(jìn)行穩(wěn)定而重復(fù)的分析�����,就需要進(jìn)行方法開發(fā)了�����。具備進(jìn)行成功方法開發(fā)的技能���,對解決將來可能遇到的復(fù)雜分析極有幫助�����。
優(yōu)化反相色譜條件
在確定了色譜柱規(guī)格�����、合適的固定相柱填料、流動相溶劑和改性劑后�����,即可開始優(yōu)化方法�����。方法的優(yōu)化是根據(jù)最終目標(biāo)決定的�����。如果要開發(fā)質(zhì)量控制方法�����,您可能會需要像等度分離這種可變因素較少的方法。如果目標(biāo)是得到最高分離度���,節(jié)省分析時間并不是首要問題���,可以選擇長柱使所有組分都得到最大分離度。如果分離速度很重要���,則應(yīng)使用短柱和高流速���。對于含許多保留不同的目標(biāo)化合物的復(fù)雜樣品,用等度分離可能不能解決問題���,應(yīng)開發(fā)并優(yōu)化梯度方法���。
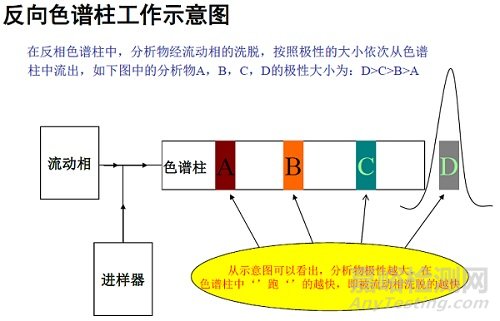
我們按約定俗成將水相溶劑和反相色譜中較弱的溶劑稱為流動相“A”,“B”溶劑是有相組成較高和較強(qiáng)的溶劑�。在反相色譜模式中,當(dāng)增加 B 的百分比時���,通常保留將減少�����。請注意�����,在還沒有開發(fā)出樣品的實際分析方法前�����,大部分人開發(fā) HPLC 方法都是采用“反復(fù)實驗”的方法���,即���,嘗試各種不同流動相條件�,以找出最佳條件。
等度(流動相組成不變)
等度優(yōu)化的一般方法是不斷改變流動相強(qiáng)度(%B)���,直到獲得適當(dāng)?shù)谋A舴秶?��。這種方法有時也稱為“溶劑篩查”。對于簡單的分離�����,k 值應(yīng)在 1 到 10 之間。如果 k 值太低(比如���,< 1)�,早洗脫的色譜峰可能不發(fā)生保留峰或融入基質(zhì)組分�,因此對其進(jìn)行定量將非常困難而且不可重復(fù)。另外�����,低 k 值峰受柱外效應(yīng)影響很大�,可能會使峰展寬。如果 k 值太大(比如�����,> 10)���,分析時間將變得過長�,檢測限會因峰展寬而提高�����。
對于反相色譜中的等度方法開發(fā)是從流動相有機(jī)改性劑最高比例開始�,然后逐漸降低�����。在降低流動相強(qiáng)度之前�����,先要確定所有組分都已從柱中洗出���。如果組分保留在色譜柱中,以后可能在 B 相比例較低的流動相中洗脫�,成為無法解釋的“鬼峰”。通常�,流動相以 +/-10%(如,90% B�、80% B、70% B 等)或 +/-20% 的幅度改變�����。利用一些現(xiàn)代軟件(如�,ChromSword 的 ChromSword�,ACD 的 AutoChrom),顯示保留變化曲線�����,系統(tǒng)進(jìn)行自動調(diào)節(jié),更快得到優(yōu)化條件�����。當(dāng)接近最佳等度條件時�,進(jìn)行手動調(diào)節(jié),幅度應(yīng)減為 5% 或 3%�����。
請注意�����,如果流動相“A”中含高濃度緩沖液(比如�,大于 25 mM),則不能使用 100% 的 B�����,因為當(dāng)兩種溶劑系統(tǒng)開始混合���,隨著 B 相百分比的減少�����,可能發(fā)生沉淀���。一旦建立優(yōu)化條件�,如果目標(biāo)還沒有得到完全分離���,則需繼續(xù)提高選擇性 (α)�。如需分離兩個相鄰的色譜峰會涉及到許多實驗參數(shù)的調(diào)節(jié)�����。溫度可以作為一個變量���。大多數(shù)現(xiàn)代儀器都具有某種類型的柱溫控制器,大部分反相柱可以承受高達(dá) 60 ℃ 的溫度���,有些甚至更高。一般來說���,升高溫度可以縮短所有峰的保留時間�,但對有些峰的影響可能與對其它峰不同,從而導(dǎo)致選擇性發(fā)生變化�����。
其它用于改變選擇性的變量如下:
1. pH(對于可離子化的化合物)
2. 緩沖液(離子)強(qiáng)度
3. 緩沖液類型(比如�,磷酸鹽、醋酸鹽或甲酸鹽�,取決于需要的 pH 范圍)
4. 流動相有機(jī)改性劑(比如,從乙腈變?yōu)榧状蓟騼烧叩幕旌弦?����;可使用三元和四元流動相混合溶劑?/span>
5. 流速(一般而言�,由于提高了柱效,較低的流速可能對分離略有改善�����,但也有例外——見下面的提示)
6. 離子對試劑濃度(如果使用離子對 RPC)
如果調(diào)節(jié)這些參數(shù)還不能改善分離�,那么最好的解決辦法就是改變色譜柱或固定相�����。更長的色譜柱具有更多塔板數(shù)�����,因此有助于分離,但切記�,分離度只能增加長度的平方根。柱長加倍使分析時間和溶劑消耗加倍�,而靈敏度降低���,但只能使分離度提高約 40%���。改用較小粒度色譜柱也能增加塔板數(shù)�,但柱壓上升 。使用新的固定相(如 C18 換成苯基柱)需要進(jìn)行一套新的溶劑篩選實驗�,耗費(fèi)更多時間,但可能得到最佳的分離結(jié)果�。但需要額外購買具有不同固定相的反相柱。
梯度(流動相強(qiáng)度隨時間變化)
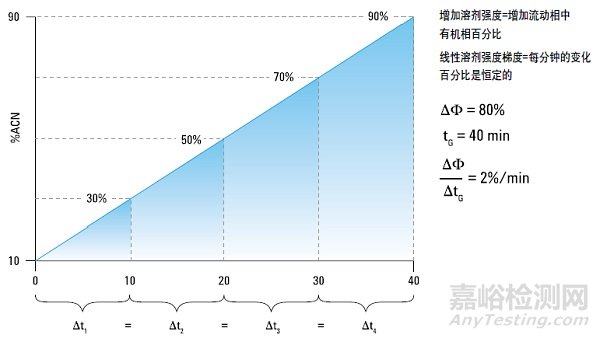
對于含各種組分的樣品混合物來說�����,選擇單一流動相組分可能得不到滿意的解決方案(比如,一般洗脫問題)�����。例如���,某些樣品組分,如強(qiáng)極性分析物�,可能很快從反相柱上被洗脫下來,而疏水性組分可能在疏水性的 C8 或 C18 上吸附非常強(qiáng)���,洗脫不下來�����。對這一問題的解決方案是隨時間改變流動相組成(梯度洗脫)���。在絕大多數(shù)例子中,初始流動相非常弱(比如���,含水量高),隨時間的推移�,有機(jī)溶劑的百分比通常是以線性形式增加
兩種情況下要使用梯度洗脫的方法開發(fā)過程���。一種是用梯度預(yù)測反相分離的最佳起始等度條件。大多數(shù)方法開發(fā)軟件(如�,DryLab、ChromSword�����、AutoChrom)都具有這項功能�����,最少進(jìn)行兩次梯度分離�,然后預(yù)測出最佳等度條件。接下來就可以用這些條件進(jìn)一步進(jìn)行等度分離優(yōu)化了�����。
第二種情況是開發(fā)梯度方法�,并使用了寬范圍的快速梯度(比如,10 分鐘內(nèi) B 由 5% 變到 95%)�����,確定目標(biāo)化合物的最佳梯度范圍�����。實際上,某些軟件系統(tǒng)可以通過色譜數(shù)據(jù)系統(tǒng)/控制器之間的相互作用來設(shè)置最佳梯度�,對分離進(jìn)行優(yōu)化。但是�����,手動操作也可以完成同樣的工作�����,找到目標(biāo)峰洗脫的流動相比例���,然后通過下一次梯度,調(diào)節(jié) k*(梯度等于 k)范圍�,讓梯度分離在合理的時間內(nèi)完成。
然后�����,與等度優(yōu)化一樣�����,當(dāng)分離時間合理后進(jìn)行選擇性的確定。
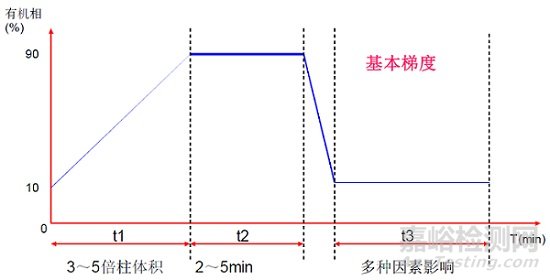
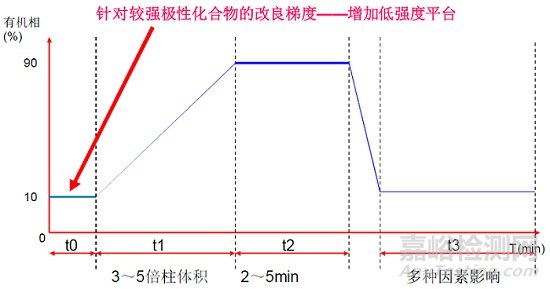
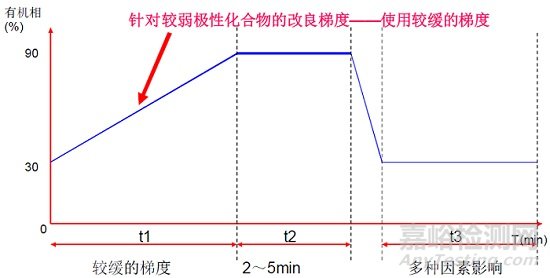
幾種常規(guī)的梯度設(shè)置
HILIC色譜柱
親水相互作用液相色譜 (HILIC) – 有時也被稱為“水相正相”(ANP) – 是一項已發(fā)展了數(shù)十年的技術(shù)�。由于這項技術(shù)能夠?qū)Ψ聪嘀喜槐A艋虮A糨^差的極性化合物進(jìn)行分析,近年來又重新受到了重視���。而且�����,這項技術(shù)很容易與 MS 和 MS-MS 檢測技術(shù)兼容�����。當(dāng) HILIC 分離使用高有機(jī)相流動相時���,與水相緩沖系統(tǒng)相比由于具有較低的離子抑制,從而提高了 MS 靈敏度�����。
HILIC 使用極性固定相���,如硅膠�����、氨基�����、混合模式�、兩性離子等,以及水溶性的�����、非極性流動相�����,由少量水(至少 2.5% 體積比)和高比例有機(jī)相組成�����。
在 HILIC 方法中�,親水���、極性和帶電荷化合物比疏水�����、中性化合物更容易保留���。與反相液相色譜完全相反�。
在流動相中加水會降低保留���。HILIC 與正相色譜相比���,能得到強(qiáng)極性溶質(zhì)的良好峰形。是反相色譜的補(bǔ)充方法�,由于保留了親水性化合物,因此常使洗脫順序倒置���。
正相色譜
正相或吸附色譜的出現(xiàn)早于反相色譜�。“正”的叫法源自其原本概念���,即���,固定相為極性、流動相為非極性���、極性化合物保留更強(qiáng)�����。
在正相色譜中�����,使用硅膠或另一種極性固定相�����,如短鏈氨基或二醇�。流動相為非極性——通常為烴類、二氯甲烷�、乙酸乙酯�,或另一種水溶性的溶劑。在正相色譜中�,極性組分保留更強(qiáng)。隨流動相極性增大極性組分保留減弱�。使用的流動相極性越強(qiáng),分析物的洗脫越快���。在硅膠柱上進(jìn)行正相梯度分離時�����,可以從己烷等非極性溶劑開始�����,然后逐漸加入極性溶劑�,如乙酸乙酯,比如�,乙酸乙酯從 5% 到 95%。根據(jù)目標(biāo)物的洗脫位置�����,可以調(diào)節(jié)梯度以改善所有組分的分離�。有時加入一定量的水或少量異丙醇,可以調(diào)節(jié)硅膠填料的表面活性�。鍵合相色譜柱可能不需要水的存在,并常使用醇類作為改性劑�。
我們使用正相色譜的原因之一是使更多的極性化合物得到保留。也可以用這種模式洗脫在反相色譜中高度保留的疏水化合物���。
正相色譜有許多用途���。適合分離幾何異構(gòu)體和位置異構(gòu)體。可得到比反相色譜更高的分辨率�。當(dāng)我們沒有水溶性的溶劑時,可以使用正相色譜�����。也可以用于制備分離�,流動相不含水,易于蒸發(fā)���。
總結(jié):正相
• 柱填料為極性:硅膠(最強(qiáng))>氨基>二醇>氰基(最弱)
• 流動相為非極性:己烷���、異辛烷、二氯甲烷���、乙酸乙酯等
• 極性化合物保留更強(qiáng)
• 隨流動相極性增大保留減弱
選擇正相的目的:
• 分離高度保留的疏水樣品
• 分離異構(gòu)體
• 樣品進(jìn)樣溶劑為非極性和/或非水溶性的
• 使用非極性溶劑進(jìn)行回收
離子交換色譜
離子交換色譜可以分離離子型化合物和可離子化的化合物���。在這種模式中���,我們使用帶有離子型官能團(tuán)的填料�,其所帶電荷與分析物相反�。在強(qiáng)陽離子交換 (SCX) 色譜中,我們要分析帶正電荷的分子或陽離子,因此使用的是陰離子型或帶負(fù)電荷的固定相�。如果我們要分析帶負(fù)電荷的分子或陰離子,應(yīng)使用陽離子型或帶正電荷的固定相�。
在離子交換色譜中,流動相通常含水量較高�,并含有緩沖液或鹽。通過連續(xù)或階梯梯度增加離子強(qiáng)度(鹽濃度)進(jìn)行洗脫�����。常用于對生物大分子的分離���,也用于小分子的分離�����,如氨基酸���、無機(jī)陽離子和陰離子,以及氨類或羧酸等可離子化的化合物�����。
離子交換色譜小結(jié):
• 柱填料含離子基團(tuán)(如�����,磺酸鹽、四烷基銨)
• 流動相為水相緩沖液(如���,磷酸鹽���、甲酸鹽、TRIS 等)
• 比反相色譜用得少
• 與離子對色譜相似(更多信息請參見詞匯表)
凝膠滲透色譜/體積排阻色譜
在 GPC/SEC 中�����,樣品分子與填料之間不存在相互作用�,并通過擴(kuò)散進(jìn)入多孔聚合物基質(zhì)或硅膠基質(zhì)的孔隙中。根據(jù)分子的體積大小和相應(yīng)填料孔隙大小進(jìn)行分離�����。體積比填料孔隙大的分子不能擴(kuò)散進(jìn)入填料���,而體積比填料孔隙小的分子則可以進(jìn)入填料���,因此樣品得到分離�。與反相色譜不同的是���,大分子先洗脫出來,小分子后洗脫出來���。
通常�����,較大分子被排阻在孔隙之外�,從而快速從色譜柱中洗脫出來���,為完全排阻體積�。最小的分子可以滲透入色譜柱的所有孔隙�,最后洗脫出來,為完全滲透體積���。其它所有分子在這兩者之間按分子大小洗脫出來�����。如果要測定單個組分的分子量 (MW) 或整體分布�����,需要先用已知分子量的標(biāo)樣以洗脫體積對 log MW 建立校正曲線�。然后,在與標(biāo)樣相同的條件下分析聚合物樣品�,即可測定該聚合物的分子量和分子量分布。
主要選用流動相溶解分析物�����。
GPC/SEC 小結(jié):
• 兩種模式:非水相 GPC 和水相 SEC(也稱凝膠過濾色譜�,或 GFC)
• 在體積排阻色譜中,樣品分子與填料之間無相互作用���,并通過擴(kuò)散進(jìn)入多孔聚合物基質(zhì)的孔隙中�。根據(jù)分子的體積大小和相應(yīng)填料孔隙大小進(jìn)行分離�。在不同溶劑或流動相中可能得到不同的流體動力學(xué)半徑。
• 主要選用流動相溶解分析物
• 主要用于聚合物表征�����、聚合物分子量測定和蛋白質(zhì)分離