摘要: 消毒劑消毒程序及驗證是GMP 體系的重要技術(shù)要求�,既是藥品生產(chǎn)企業(yè)落實GMP 的重要手段,也是GMP 現(xiàn)場檢查的關(guān)注重點���。消毒劑消毒效力驗證應(yīng)結(jié)合企業(yè)實際使用情況���,《消毒技術(shù)規(guī)范》���、歐洲標準EN13697:2015 等國內(nèi)外消毒劑檢測相關(guān)法規(guī)主要針對的是消毒劑產(chǎn)品本身���,而消毒劑的消毒效力還需要考慮企業(yè)自身的應(yīng)用場景和方式,如消毒劑接觸材質(zhì)�、消毒方式(擦拭、噴灑或浸泡等)�、消毒操作的一致性�����、作用時間以及環(huán)境中常見微生物等相關(guān)因素���。建議企業(yè)在結(jié)合自身消毒工藝的基礎(chǔ)上,參考國內(nèi)外消毒劑行業(yè)相關(guān)標準和指南�����,制定適配的消毒劑消毒效力驗證方案�����。
消毒劑(disinfectant)是一類用于將活性微生物數(shù)量降至安全水平的化學試劑���,為潔凈區(qū)環(huán)境控制提供關(guān)鍵保障�����。在制藥行業(yè)中,消毒劑及其使用程序的有效性必須通過嚴格的驗證來證實���,以確保其能從各種材質(zhì)表面有效去除微生物�����。盡管消毒劑廠商會提供產(chǎn)品數(shù)據(jù)�����,但藥品生產(chǎn)企業(yè)仍需自主完成相應(yīng)驗證�����,以確保消毒劑的殘留物不影響任何微生物的檢出[1]�����。
當前���,在推進與國際主要監(jiān)管機構(gòu)接軌的過程中�,需深入分析新修訂的PIC/S GMP 附錄1[2]���、FDA 行業(yè)指南(Guidance for Industry)及EU GMP 中關(guān)于消毒劑效力驗證的技術(shù)要求差異���,同時參照PDA[3]、ASTM[4]等標準,系統(tǒng)研究檢測方法和技術(shù)指南���。在此基礎(chǔ)上���,企業(yè)可進一步完善消毒劑效力驗證體系,監(jiān)管機構(gòu)可加強相關(guān)監(jiān)管工作�����,共同提升藥品生產(chǎn)質(zhì)量管理水平�����。
1 潔凈區(qū)消毒劑的選擇及其檢查要點
評估藥品生產(chǎn)企業(yè)所用的消毒劑和現(xiàn)有消毒規(guī)程�,應(yīng)重點關(guān)注企業(yè)是否選用了適宜的消毒劑,并對消毒劑的輪換使用周期和種類做出了具體規(guī)定�,例如在不同潔凈級別的區(qū)域是否選用對應(yīng)風險等級的消毒劑,以及各種類消毒劑在作用機制上是否存在差異���;所選消毒劑是否具備廣譜殺菌能力���、低殘留和低腐蝕性的特點;尤其在A/B 級潔凈區(qū)�����,因污染控制要求極高���,必須使用無菌型消毒劑���;同時,應(yīng)選用有效的殺菌劑和殺真菌劑�����,并重視殺孢子劑的使用�,以確保對潔凈區(qū)內(nèi)細菌和真菌都具有殺滅效果。
消毒劑選擇的檢查要點包括:① 對于成品包裝消毒劑�,藥品生產(chǎn)企業(yè)應(yīng)在選定供應(yīng)商合作初期,進行審計�����、核查其提供的研究資料是否完備�,包括詳細信息、材料相容性�、腐蝕性等基礎(chǔ)研究數(shù)據(jù),以及消毒劑的儲存條件���、銷毀方式�����、包裝形式���、無菌等相關(guān)信息�����。若企業(yè)采用自配消毒劑�,應(yīng)通過全面風險評估的形式�����,確定科學的管理策略及潛在風險���,同時須進行殺菌效力驗證�����。若涉及的自配消毒劑需除菌過濾���,還應(yīng)進行無菌驗證�����。② 依據(jù)GMP 對物料管理的相關(guān)規(guī)定�,無菌消毒劑入廠驗收的相關(guān)流程應(yīng)規(guī)范化�����。企業(yè)應(yīng)明確規(guī)定消毒劑的入廠檢測�、放行出庫�����、使用銷毀的全生命周期管理要求���。③ 若將消毒劑作為“成品”���,需要考慮其容器的選擇,如無菌型消毒劑噴壺是否為無菌噴壺���,在使用過程中是否保持無菌�。④ 在A/B 級潔凈區(qū)中的潔凈工具(如拖布���、擦拭布)使用前是否確定無菌�����。⑤ 外購商品化無菌型消毒劑是否有輻照證書���、輻照驗證報告和放行的無菌檢測報告�����。⑥ 企業(yè)自行過濾消毒劑的無菌過濾系統(tǒng)須經(jīng)過驗證�,濾芯應(yīng)進行無菌性和完整性測試�����,并重點關(guān)注濾芯和消毒劑的兼容性�����、吸附性�����、析出情況及菌體截留效果�����、過濾前后的有效成分變化及無菌達標情況等。⑦ 企業(yè)應(yīng)定期使用殺孢子劑�,且所選消毒劑種類應(yīng)涵蓋低、中�、高效力范圍。不同級別的潔凈區(qū)和設(shè)備使用殺孢子劑的頻次及種類也需要密切關(guān)注�����。⑧ 消毒劑開瓶有效期的考察�����。在實驗室驗證部分模擬真實場景的保存區(qū)域和開瓶方式���,若消毒劑存在配制前后母液及工作液的保存,應(yīng)同步考察���,通過每日模擬實際使用�����,至規(guī)定效期時進行殺滅驗證�。⑨ 自配型消毒劑配制方法的確認。明確消毒劑名稱�、各成分用量及最終體積,驗證配制的消毒劑有效成分濃度符合要求���,確保自配消毒劑質(zhì)量穩(wěn)定可靠�。
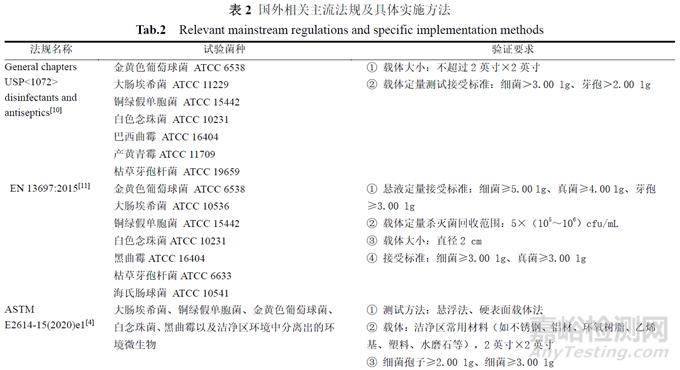
消毒劑的殘留問題亦是GMP 檢查過程中的關(guān)注重點���,主要包括:消毒劑生產(chǎn)廠家提供殘留報告的���,需重點考察殘留的可接受標準以及廠家報告的殘留測試方式;企業(yè)需要確認消毒劑是否有殘留�����,殘留是否有潛在危害�����,并制定去除殘留的措施�����。在消毒SOP 中應(yīng)規(guī)定殘留清除操作要求���,如作用一定時間后再用滅菌的純化水�����、注射用水或70%異丙醇擦拭�。此外,對GMP 指南 �、FDA 行業(yè)指南(Guidance for Industry) 和EU GMP 中主要檢測法規(guī)的拆解顯示:FDA 行業(yè)指南中規(guī)定消毒劑和消毒程序的有效性應(yīng)以其從表面充分去除潛在污染物的能力來衡量,尤其對于一部分不規(guī)則表面�,由于其形狀復(fù)雜,難以清潔和消毒���,容易成為微生物滋生的溫床。FDA 強調(diào)���,消毒劑效力驗證須涵蓋所有關(guān)鍵工藝和生產(chǎn)區(qū)域中使用的載體�,包括不規(guī)則表面���,并需要評估消毒劑在不同表面上的殺菌效力及接觸時間���,確保其能夠達到預(yù)期的殺菌效果。這說明實驗室驗證中載體定量殺滅試驗數(shù)據(jù)結(jié)果的充分性對于清潔消毒體系的建立具有舉足輕重的作用[5]�。EU GMP 同樣要求驗證消毒劑在有效期內(nèi)的有效性���,并應(yīng)考慮其接觸時間、使用方式和其接觸的表面材質(zhì)[6]���。因此���,接觸時間、方式及材質(zhì)的選擇均為此項驗證中的重點考量范疇���。對于消毒劑效力驗證試驗�,不同國家或組織的相關(guān)主流法規(guī)及具體實施方法的檢查重點見表1�、表2。
從上述表格中可知�,國內(nèi)外相關(guān)主流法規(guī)及具體實施方法的差異,具體如下:
① 各法規(guī)中菌株種類基本一致���,其中美國法規(guī)要求中增加了產(chǎn)黃青霉�,歐盟法規(guī)中增加了海氏腸球菌���;
② 中美歐對載體大小的要求均有不同���,其中中國國標要求的載體大小最?����?;
③ 殺滅判定結(jié)果�,懸液法的判定標準以歐盟和中國的《消毒技術(shù)規(guī)范》要求最為嚴苛;
④ 菌液初始接種量存在細微的差異�;
⑤《藥品微生物實驗室消毒劑效力評估指導(dǎo)原則》則簡化了有效性和無毒性兩方面的驗證,引入了清晰�����、量化的驗收標準(回收值為0.5~2.0 倍)�,并強調(diào)了使用低濃度微生物(10~102 cfu/mL)開展驗證。
結(jié)合國內(nèi)外相關(guān)法規(guī)的實施要點�����,為消毒劑效力驗證的檢查要點制定提供了許多啟發(fā)�。具體需要關(guān)注以下幾個方面:
① 標準菌株的涵蓋范圍:驗證所用標準菌需包括細菌���、酵母菌和霉菌的系統(tǒng)范疇�����。針對不同類型的消毒劑對應(yīng)不同的標準菌株���,如低效消毒劑針對金黃色葡萄球菌(革蘭陽性菌代表)�����、大腸埃希菌或者銅綠假單胞菌(革蘭陰性菌代表)���、白色念珠菌(酵母菌及真菌代表);中效消毒劑在此基礎(chǔ)上可適當增加黑曲霉(產(chǎn)孢霉菌代表)�����;殺孢子劑則選用枯草芽孢桿菌或者枯草桿菌黑色變種芽孢(芽孢桿菌代表)�����。
② 環(huán)境菌株的納入考量:菌株選擇中是否考慮加入環(huán)境菌的相關(guān)要求���,可依據(jù)消毒劑特殊用途或者試驗需求���,結(jié)合年度微生物鑒定回顧及污染事件中的典型菌種�,適當增選環(huán)境代表菌株�����。針對環(huán)境菌的篩選應(yīng)符合高頻次�����、多種類的原則���。殺孢子劑應(yīng)用時���,對環(huán)境菌的檢測范圍還需涵蓋環(huán)境中的典型芽孢及真菌孢子。
③ 消毒劑作用時間的確定:消毒劑需經(jīng)過篩選與載體定量殺滅試驗結(jié)果進行確定���,驗證方式需結(jié)合法規(guī)推薦的相關(guān)要求進行參考���。
④ 中和劑的合理選擇:所選中和劑應(yīng)既能有效抑制微生物生長不受干擾,又能高效中和消毒劑中的化學成分�,從而能準確地評估消毒劑在特定時間內(nèi)的真實殺菌效果。
2 消毒劑效力驗證中關(guān)于材質(zhì)選擇的檢查重點
載體定量殺滅試驗是用來評估消毒劑和殺孢子劑對微生物的殺菌效果���,需在不同類型材質(zhì)上進行模擬測試[11-12]。應(yīng)選擇藥品生產(chǎn)企業(yè)實際場景中具有代表性的表面材質(zhì),包括常見材質(zhì)�、易殘留污染物的多孔性載體或較難清潔的典型材質(zhì)(如不銹鋼、塑料���、塑料袋�、玻璃���、軟簾�、聚碳酸酯)�、不同的地板材料(如環(huán)氧樹脂、乙烯)�、墻壁材料(如環(huán)氧涂層)等。材質(zhì)種類的選擇應(yīng)依據(jù)其表面附著微生物后對最終產(chǎn)品帶來的風險水平���,這類表面可認為是關(guān)鍵表面���。以不銹鋼為例,盡管其表面光潔且易于清洗�,但它可能是直接接觸產(chǎn)品的表面材質(zhì),從這個角度而言�,對最終產(chǎn)品質(zhì)量影響風險最大,因而被判斷為關(guān)鍵表面�����。
風險等級判定、材質(zhì)占比判定�����、清潔標準判定參考以下原則:
① 低占比材質(zhì)�����。在潔凈區(qū)鮮有出現(xiàn)�,一般位于某些設(shè)備的輔助細節(jié)小面積區(qū)域或者工器具的小面積輔助材質(zhì)。
② 中等占比材質(zhì)�。在潔凈區(qū)較為常見,一般出現(xiàn)在某些設(shè)備的主要部位或者工器具的基本材質(zhì)�。
③ 高占比材質(zhì)。在潔凈區(qū)最為常見�����,一般為整體潔凈區(qū)域的大面積墻面���、地面�����、天花板以及主要設(shè)備�����、桌面�����、凳子和小推車等的主要材質(zhì)表面���,或者大型工器具的主要材質(zhì)。
清潔標準判定依據(jù):
① 低風險區(qū)域的清潔要求�。如C/D 級潔凈區(qū)中無關(guān)鍵操作區(qū)域?qū)?yīng)的材質(zhì)。
② 中等風險區(qū)域的清潔要求�����。如C/D 級潔凈區(qū)中有關(guān)鍵操作區(qū)域?qū)?yīng)的材質(zhì)�����,或B 級潔凈區(qū)中無關(guān)鍵操作區(qū)域?qū)?yīng)的材質(zhì)�����。
③ 高風險區(qū)域的清潔要求。A 級區(qū)域所有材質(zhì)�,以及B 級潔凈區(qū)中關(guān)鍵操作區(qū)域?qū)?yīng)的材質(zhì)。
藥品生產(chǎn)企業(yè)可結(jié)合自身潔凈區(qū)所使用的材質(zhì)種類和消毒劑類型進行風險評估�,確定出載體定量殺菌試驗對應(yīng)的測試載體。若選用有涂層的材料�����,應(yīng)確保涂層穩(wěn)固�、無脫落或吸附等問題,以免干擾試驗結(jié)果�。在生產(chǎn)區(qū)域,不同接觸表面的材質(zhì)各異�,設(shè)備包括不銹鋼、聚四氟乙烯�����、鋼化玻璃等�����;手套消毒常用橡膠類材質(zhì)(如乳膠���、丁腈手套)�����,隔離器手套多為氯磺化聚乙烯�����,工作鞋則為橡膠或塑料等材質(zhì)�����。這些常見且關(guān)鍵材質(zhì)多為驗證代表材質(zhì)�����。此外���,在載體的來源方面,應(yīng)關(guān)注是否來自車間內(nèi)關(guān)鍵表面相對應(yīng)的供應(yīng)商�,是否具備材質(zhì)證明等相關(guān)文件。以上均需要在評估中綜合分析�����,得出最終的評估報告[13]�����。
3 消毒劑效力驗證中關(guān)于消毒作用方式的檢查重點
按照日常消毒作用方式的情況進行驗證,才能保證實驗室試驗與現(xiàn)場試驗的一致性�����。日常消毒作用方式基本可分為以下幾種:
① 潔凈擦拭布浸潤消毒劑后進行擦拭(墻面地面拖拭)或預(yù)靜置成品擦拭布擦拭���,通過從上至下或者從里至外等擦拭路徑進行擦拭���;
② 消毒劑噴壺噴灑后用潔凈擦拭布進行擦拭;
③ 單純消毒劑噴灑作用�;
④ 浸泡(常見于液封消毒、地漏的消毒)���,在實際操作中�,噴灑�����、擦拭(拖拭)�����、浸泡等清潔方式存在單一使用或多種方法協(xié)同并用的情況。
擦拭動作的規(guī)范性和細節(jié)直接影響消毒效果和污染控制�����,擦拭方向與順序是較為核心的兩個方面�����,建議單向有序擦拭�����,采用單向擦拭(如“S”型或直線型)�,避免來回反復(fù)擦拭導(dǎo)致污染物擴散�。擦拭不同區(qū)域或設(shè)備時,為避免交叉污染�����,需更換干凈的擦拭布���,或按“清潔→消毒”分步操作�����。擦拭工具應(yīng)使用無脫落纖維的無塵布(如聚酯纖維)���、一次性消毒濕巾或?qū)S脻崈裟ú?��。折疊方法為:將擦拭布折疊成4~8 層,每擦拭一個表面后翻面�,確保始終使用干凈面接觸清潔區(qū)域。浸漬量應(yīng)控制在消毒劑浸濕擦拭布至微潤且不滴水(過度濕潤可能殘留液體���,滋生微生物)�����。由內(nèi)向外�����、自上而下進行擦拭�,從潔凈度要求高的區(qū)域(如設(shè)備內(nèi)部)向較低區(qū)域擦拭�;從頂部(天花板、設(shè)備頂部)向底部(地面)進行���,避免二次污染�����,且應(yīng)覆蓋全面�����,確保擦拭面完全重疊(建議重疊1/3 寬度)�����,不留死角�����。還應(yīng)適當對擦拭材質(zhì)施加均勻壓力(以肉眼可見擦拭布與表面充分接觸為宜)���,確保消毒劑與表面充分接觸,但需避免摩擦產(chǎn)生顆粒�。消毒劑需在表面停留規(guī)定時間,擦拭后不可立即擦干(動作連貫�,避免消毒劑在擦拭過程中過早干燥,可分段操作�����,確保每段保持濕潤時間)。擦拭至區(qū)域邊緣時�,將污染物向未清潔區(qū)域方向收攏,避免將其帶回已清潔區(qū)域�。這些擦拭的關(guān)鍵點在消毒劑效力驗證時,做到完全協(xié)調(diào)統(tǒng)一較為困難�����,其實際運用價值在消毒效果現(xiàn)場測試部分更為凸顯�。
在消毒劑效力驗證實驗室的載體殺滅測試環(huán)節(jié),其作用方式考量需兼顧實際應(yīng)用場景與極端條件模擬的雙重維度�����,既需貼近真實使用環(huán)境�����,又需構(gòu)建更具挑戰(zhàn)性的"最差情況"模型以驗證消毒效力�。由于實際運用的擦拭方式較多,在實驗室尺寸有限的材質(zhì)表面進行載體殺滅測試的運用中�,難以確保操作人員的手法完全協(xié)調(diào)統(tǒng)一。在選擇最終殺滅作用接觸方式時���,考慮到用最差條件(如消毒部位結(jié)構(gòu)復(fù)雜�����、表面材質(zhì)特殊���、最難殺滅微生物種類等)去模擬的必要性�����,且擦拭在理論意義上存在一定的物理作用�����,更利于細菌的清除和殺滅���。然而,噴灑方式仍存在較差狀態(tài)的可能性�����,由于噴灑時�,霧粒分布更為隨機且分散���,同時霧粒會因蒸發(fā)或飄散而具有失效的風險�����,且隨作用力的影響較為顯著�,如壓力適中的噴壺可產(chǎn)生均勻霧粒,而高壓噴壺可能生成更細顆粒���。在潔凈區(qū)操作時���,受到潔凈環(huán)境的影響也較為普遍,如在強通風或開放環(huán)境中噴灑���,霧粒易被吹散�����,更易降低表面附著率���,高溫或低濕度環(huán)境可能導(dǎo)致霧粒快速蒸發(fā)�。粒徑在50~150μm 之間時分散較為均勻。粒徑過?����。?lt;50 μm)時,易形成氣溶膠�����,長時間懸浮在空氣中���,容易隨風擴散�����,降低表面消毒效果�。粒徑過大(>150 μm)時�,沉降速度過快,覆蓋面積受限�,可能導(dǎo)致局部液體堆積,浪費消毒劑且難以均勻覆蓋[14]�����。綜合這些影響因素���,推測噴灑方式較為隨機���、不可控、效果更差���。針對不同的應(yīng)用場景�,若使用不同作用方式的消毒劑�,建議分別進行模擬驗證。如需選擇���,應(yīng)開展充分的風險評估���,并根據(jù)評估結(jié)論實施驗證,以證明消毒劑的有效性[15]���。驗證時�����,建議噴灑量盡量低于實際使用量�,以覆蓋菌膜即可���。使用噴壺前���,宜在空曠區(qū)域測試霧粒分布�����,調(diào)整噴灑距離和角度���。噴灑遵循“濕潤不流淌”原則,確保表面均勻濕潤但無液體滴落���,確保有效接觸時間后�����,方可獲取真實的殺滅效果�����。
4 消毒劑效力驗證現(xiàn)場試驗的檢查重點
現(xiàn)場測試應(yīng)嚴格遵循現(xiàn)行清潔消毒流程�,嚴格按照既定的清潔方式�����、消毒方法、作用時間及周期標準執(zhí)行�。測試重點在于潔凈區(qū)微生物能否穩(wěn)定控制,同時需要持續(xù)關(guān)注消毒劑對消毒對象可能造成的實際腐蝕性和關(guān)鍵操作環(huán)節(jié)中消毒劑殘留的問題�����。在消毒劑效力驗證實驗室階段完成后�,所累積的載體表面實際測試結(jié)果通常能在一定程度上反映現(xiàn)場消毒效果�����,日?����?山柚h(huán)境微生物檢測數(shù)據(jù)持續(xù)監(jiān)測���。在廠房設(shè)施首次啟用或停機重啟等關(guān)鍵節(jié)點開展環(huán)境監(jiān)測���,更能直觀展現(xiàn)消毒劑的實際消毒效能。消毒劑效力受作用時間���、濃度�����、溫度���、濕度和風速等綜合因素影響���,企業(yè)需緊密結(jié)合環(huán)境監(jiān)測數(shù)據(jù),全面�、動態(tài)地評估消毒劑在實際應(yīng)用中的效果,持續(xù)動態(tài)優(yōu)化消毒流程和參數(shù)���,切實降低微生物污染和交叉污染風險�����。
藥品生產(chǎn)企業(yè)應(yīng)運用消毒劑風險評估工具�、材質(zhì)選擇的標準化思路���,并參考中國GMP���、EU GMP[16]和FDA 的在菌種管理方面的協(xié)調(diào)統(tǒng)一,交叉運用最前沿的法規(guī)的核心優(yōu)勢���,開展消毒劑效力驗證工作���。當企業(yè)在新增消毒劑廠家或者種類時�����,可在局部區(qū)域進行試用,并結(jié)合環(huán)境監(jiān)測數(shù)據(jù)評估其現(xiàn)場消毒效果���,確保驗證通過及安全可靠后再全面推廣�。消毒劑效力驗證并非一次性工作�����,當環(huán)境中出現(xiàn)耐受菌或者微生物異常調(diào)查與消毒劑效力存在相關(guān)時���,應(yīng)迅速啟動消毒劑效力確認工作�,排查問題根源�����。制藥行業(yè)在起草消毒劑效力驗證方案時�����,應(yīng)遵循監(jiān)管要求,確保所使用的消毒劑在潔凈環(huán)境中能有效控制微生物污染�。消毒劑效力驗證是制藥行業(yè)微生物管理的核心環(huán)節(jié),更是確保產(chǎn)品安全性和質(zhì)量的基石�。企業(yè)應(yīng)建立涵蓋實驗室預(yù)驗證、現(xiàn)場模擬試驗���、日常監(jiān)測和年度回顧的全生命周期驗證體系�,通過分階段評估消毒劑對目標微生物的殺滅效力�����、作用時間及環(huán)境適應(yīng)性���,持續(xù)優(yōu)化消毒程序的有效性���,確保全過程的科學性、合規(guī)性和實際適用性�。
5 結(jié) 語
消毒劑效力驗證的檢查范圍涵蓋“人、機�����、料、法���、環(huán)�����、測”六大核心要素�����,具體包括驗證人員、儀器設(shè)備���、各類物料(消毒劑�����、中和劑���、菌種、中和產(chǎn)物�、稀釋劑、培養(yǎng)基�、載體材質(zhì))���、驗證項目(中和劑鑒定試驗、懸液殺滅�、載體殺滅、效期驗證�、現(xiàn)場殺滅效果測試)、驗證環(huán)境(實驗室���、現(xiàn)場)以及檢測方法���。通過全面風險評估,識別風險點并確定等級�,為后續(xù)效力驗證方案制定提供理論支持。消毒劑效力驗證是企業(yè)內(nèi)部清潔消毒體系建立的前置條件���,應(yīng)具有專屬性���。企業(yè)選取商品化的消毒劑后,應(yīng)根據(jù)廠家提供資料的規(guī)范程度進行評估�����,確認其驗證過程和結(jié)果的適用性���,并結(jié)合企業(yè)實際開展更全面和適宜于生產(chǎn)實際情況的驗證�����,不得將廠家的驗證報告直接替代企業(yè)內(nèi)部驗證���。企業(yè)內(nèi)部自主或委托有資質(zhì)的第三方完成驗證�����,結(jié)果符合可接受標準后���,應(yīng)將經(jīng)驗證的消毒措施轉(zhuǎn)化為消毒標準操作規(guī)程或消毒管理程序,文件應(yīng)與實際操作一致�。此外�,建議在消毒劑效力驗證方案中明確規(guī)定再驗證觸發(fā)條件,包括但不限于:新增廠區(qū)或新增需清潔消毒的潔凈區(qū)域�,更換消毒劑廠家或消毒劑種類、濃度等發(fā)生變化���,環(huán)境監(jiān)測中出現(xiàn)首次驗證未覆蓋的非代表菌屬污染���,以及任何可能影響潔凈區(qū)清潔消毒效果的關(guān)鍵參數(shù)變化���。出現(xiàn)上述任意情形時,均應(yīng)按照變更控制程序啟動再驗證�����。同時�,探討消毒劑效力驗證的生產(chǎn)現(xiàn)場檢查關(guān)注策略,可以更好地為藥品生產(chǎn)企業(yè)GMP 檢查提供新的思路和參考依據(jù)�。
參考文獻
[1] 國家藥典委員會. 中華人民共和國藥典四部[M]. 北京: 中國醫(yī)藥科技出版社, 2025: 725-727.
[2] PIC/S Secretariat. Important factors in validation of aseptic manufacturing[S/OL]. Geneva: PIC/S. (2011-01-01)[2025-10-08].
https://picscheme.org/docview/3446.
[3] PDA. Fundamentals of cleaning and disinfection programs for aseptic manufacturing facilities[S/OL]. Bethesda: PDA. (2015-07-01)[2025-10-08]. https://www.pda.org/bookstore/product-detail/2809-tr-70-cleaning-and-disinfection-programs.
[4] ASTM International. E2614-15(2015). Standard guide for evaluation of cleanroom disinfectants [S]. West Conshohocken: ASTM International, 2015.
[5] FDA. FDA-2003-D-0145. Sterile drug products produced by aseptic processing-current good manufacturing practice[S/OL]. Silver Spring: FDA . (2004-10-01)[2025-10-08]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/sterile-drug-products-produced-aseptic-processing-current-goodmanufacturing-practice.
[6] European Commission. Qualification and validation: Annex 15 to the EU GMP guide [S/OL]. Brussels: European Commission. (2015-03-30) [2025-10-08].
https://health.ec.europa.eu/system/files/2016-11/2015-10_annex15_0.pdf.
[7] 高東旗. 國家衛(wèi)生部頒布新的《消毒技術(shù)規(guī)范》[J]. 醫(yī)學動物防制, 2003,19 (6): 384.
[8] 國家市場監(jiān)督管理總局. 消毒劑實驗室殺菌效果檢驗方法. GB/T 38502-2020 [S/OL]. 北京:國家市場監(jiān)督管理總局. (2020-03-06) [2025-10-08].https://openstd.samr.gov.cn/bzgk/gb/newGbInfohcno=744F030ACEF7928C91AE0CF5C2F306AE.
[9] 國家市場監(jiān)督管理總局. 噴霧消毒效果評價方法. GB/T 38504-2020 [S/OL]. 北京:國家市場監(jiān)督管理總局. (2020-03-06)[2025-10-08]. https://openstd.samr.gov.cn/bzgk/gb/newGbInfo?hcno=63620CECE811DE773847B9AC79E9006E.
[10] United States Pharmacopeial Convention. General chapters USP<1072> disinfectants and antiseptics [S]. Maryland: United States PharmacopeialConvention, 2017.
[11] European Committee for Standardization. EN 13697:2015. Chemical disinfectants and antiseptics-quantitative non-porous surface test for the evaluation ofbactericidal and/or fungicidal activity of chemical disinfectants used in food, industrial, domestic and institutional areas-test method and requirements withoutmechanical action (phase 2, step 2)[S]. Brussels: European Committee for Standardization. 2015.
[12] U.S. Environmental Protection Agency. Methods and guidance for testing the efficacy of antimicrobial products against spores of Clostridioides difficile onhard non-porous surfaces[S/OL]. Washington: EPA. (2022-09-01)[2025-10-08]. https://www.epa.gov/pesticide-registration/methods-and-guidance-testingefficacy-antimicrobial-products-against-spores.
[13] DI MARTINO G, PASQUA S, DOURADINHA B, et al. Efficacy of three commercial disinfectants in reducing microbial surfaces' contaminations ofpharmaceuticals hospital facilities[J]. Int J Environ Res Public Health, 2021, 18(2): 779.
[14] 李炎, 李陸瑤, 段弘揚, 等. GB/T 38504-2020《噴霧消毒效果評價方法》標準解讀[J]. 中國消毒學雜志, 2022, 39(8): 622-624.
[15] WILLISON-PARRY D, YANG S, FORNG R Y, et al. Disinfectant efficacy: understanding the expectations and how to design effective studies that includeleveraging multi-site data to drive an efficient program[J]. PDA J Pharm Sci Technol, 2020, 74(2): 249-263.
[16] European Commission. EU GMP Annex 1: manufacture of sterile medicinal products[EB/OL]. (2022-08-22)[2025-10-08].https://health.ec.europa.eu/system/files/2022-08/20220825_gmp-an1_en_0.pdf.