抗體藥物偶聯(lián)物(ADC)是兩種不同類型分子的偶聯(lián)物,常見組合是一端為單抗�,另一端是小分子藥物載荷。因此�,ADC的非臨床安全性評估既要考慮對ADC整體的評價,也要考慮載荷帶來的風(fēng)險�。目前專門針對ADC非臨床評價的指導(dǎo)原則不多,可以參考NMPA 2023年9月發(fā)布的《抗體偶聯(lián)藥物非臨床研究技術(shù)指導(dǎo)原則》�,也可以參照一些涉及ADC的通用型指南�,比如ICH M3(R2)�、ICH S6(R1)、ICH S9及Q&A�。結(jié)合已有指南和公開文獻,圍繞ADC這類產(chǎn)品的非臨床研究內(nèi)容做下分享�。
藥理學(xué)研究
主要藥效學(xué)研究
可通過多種體外和體內(nèi)實驗評估ADC及其組成部分的臨床前藥效學(xué)活性。大致研究內(nèi)容包括與目標(biāo)靶點的結(jié)合能力(含特異性)�、內(nèi)吞能力、靶細胞殺傷能力(ADCC/CDC/載荷毒性)�、載荷解離能力、旁觀者效應(yīng)�、組織交叉反應(yīng)、體內(nèi)抗腫瘤作用�、PK/PD等研究。文末羅列了13款已上市ADC的藥理學(xué)研究內(nèi)容�。
組織交叉反應(yīng)研究(TCR):根據(jù)ICH S9問答文件,組織交叉反應(yīng)試驗意義有限�,通常不需要開展。當(dāng)無藥理學(xué)相關(guān)種屬時�,首次人體試驗(FIH)應(yīng)考慮組織交叉反應(yīng)研究或其他替代方法,與NMPA的思路一致�。不過�,如ADCETRIS、Trodelvy�、Elahere�、TIVDAK等已上市ADC�,大都開展了TCR試驗的。
次要藥效學(xué)研究
ADC是高活性藥物�,如果單抗是新分子,建議在研發(fā)早期開展下脫靶研究�,提前排除風(fēng)險。不過�,不屬于申報強制要求內(nèi)容之列。
安全藥理學(xué)研究
ADC對心血管�、呼吸和中樞神經(jīng)系統(tǒng)功能的潛在影響通常作為標(biāo)準(zhǔn)GLP重復(fù)給藥毒性研究的一部分進行評估,即大家通常理解的伴隨開展�。心血管/呼吸評估通常在活動受限的動物中進行,包括心電圖(ECG)檢查�,測量PR間期、QRS波群�、QT間期、校正的QT間期(QTc)�,特別關(guān)注QT間期延長風(fēng)險。同時還需測量心率和血壓�。神經(jīng)學(xué)評估包括對環(huán)境刺激的反應(yīng)、眼部�、運動反射、感覺功能和體溫的評估等�。
還是以T-DM1為例,開展了雌性食蟹猴單次給藥安全藥理學(xué)研究,使用外科植入的遙測發(fā)射器進行心血管安全性評估�,以及在重復(fù)劑量研究中對相同劑量水平的心電圖和心率進行評估。在兩項研究中均未觀察到QT延長�。在遙測動物中觀察到血壓一定程度升高,但在重復(fù)給藥毒理研究中未發(fā)現(xiàn)相應(yīng)的顯微鏡下病變�。其單抗成分(曲妥珠單抗)的臨床前研究也未發(fā)現(xiàn)心臟毒性,且在患者中進行的T-DM1治療劑量的QT研究中觀察到的異常反應(yīng)極小�。然而,在需要停用T-DM1的患者中觀察到低發(fā)生率的高血壓�。接受曲妥珠單抗治療的患者發(fā)生左心室功能障礙的風(fēng)險增加,接受ADC治療的患者風(fēng)險相似�。在食蟹猴中進行的T-DM1神經(jīng)學(xué)評估中未發(fā)現(xiàn)異常。然而�,在坐骨神經(jīng)和脊髓背索中觀察到劑量依賴性的顯微鏡下軸突變性,在6周的研究恢復(fù)期內(nèi)未顯示出可逆性�。TCR研究結(jié)果顯示,在膠質(zhì)細胞和周圍神經(jīng)梭形細胞中出現(xiàn)低強度膜染色�,證實了該毒性是供試品相關(guān)的。
載荷的安全藥理學(xué):除了體外hERG試驗外�,通常不單獨對載荷進行安全藥理學(xué)評估。hERG鉀離子通道在心肌復(fù)極化中起重要作用�,其阻斷與QT延長和尖端扭轉(zhuǎn)型室性心動過速相關(guān)。由于載荷可能存在于循環(huán)中�,會檢查小分子載荷對鉀離子通道的濃度-反應(yīng)關(guān)系。對DM1載荷的hERG試驗評估發(fā)現(xiàn)�,其IC50高于受試的最高劑量水平�,比接受T-DM1的患者檢測到的DM1濃度至少高30倍�。
藥代動力學(xué)研究
ADC臨床前PK評估范圍無統(tǒng)一標(biāo)準(zhǔn)�,需基于分子特征確定。ADC由單抗�、小分子載荷和連接子組成,其臨床前PK評估內(nèi)容廣泛�。完整的ADC臨床前PK研究需通過體外和體內(nèi)實驗,分別評估單抗�、載荷(可能包括載荷-連接子片段代謝物)等單個組分,以及完整ADC的藥代動力學(xué)特征�。
盡管ADC的治療目標(biāo)之一是減少游離載荷的全身暴露,但循環(huán)中載荷的意外釋放及藥物制劑中少量游離載荷的存在難以避免�,因此需全面表征載荷的PK特征以明確潛在安全風(fēng)險。
對于新載荷�,載荷的臨床前PK表征包括大小動物PK、組織分布試驗�、排泄試驗、血漿穩(wěn)定性試驗�、體內(nèi)外代謝產(chǎn)物鑒定、體外代謝穩(wěn)定性�、CYP450相互作用(抑制、誘導(dǎo)及反應(yīng)表型分析)�、血漿蛋白結(jié)合率、外排與內(nèi)流轉(zhuǎn)運體檢測等�。若載荷已有充分研究,可參考已發(fā)表文獻中的非臨床PK數(shù)據(jù),替代或補充新增PK實驗�。可以考慮采用放射性標(biāo)記技術(shù)開展�。
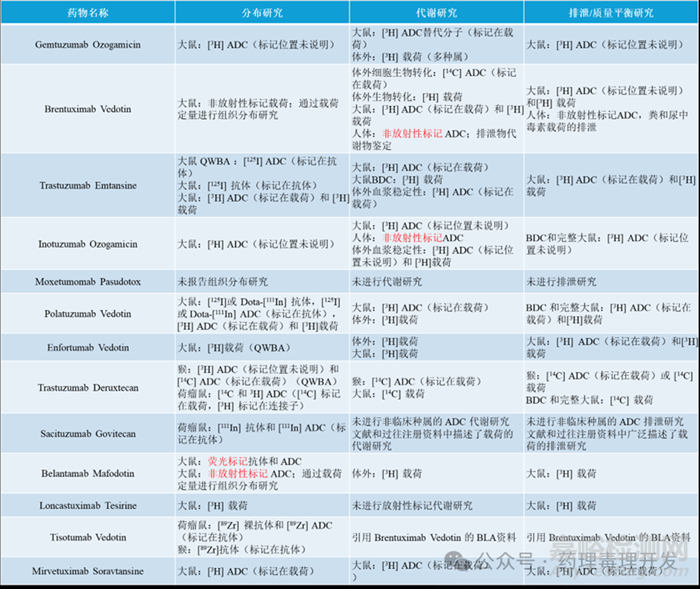
已獲批ADC產(chǎn)品開展的非臨床分布、代謝和排泄研究匯總?cè)缦拢?/span>
圍繞CYP450和轉(zhuǎn)運體相關(guān)研究額外聊幾句�。由于毒素的人體暴露量非常低,遠低于影響體外代謝酶�、轉(zhuǎn)運體的IC50,因此大部分情況下毒素分子誘導(dǎo)或抑制酶�、轉(zhuǎn)運體的風(fēng)險比較低。比如�,體外研究顯示,MMAE�、DM-1對肝藥酶CYP3A的抑制濃度在微摩爾水平,但攜帶這兩個毒素的ADC產(chǎn)品brentuximab vedotin和T-DM1臨床擬用劑量下的毒素暴露量在納摩爾級別�。實際上,brentuximab vedotin開展的臨床DDI研究顯示�,與CYP3A底物咪達唑侖聯(lián)合用藥,并未影響其暴露量�,與體外數(shù)據(jù)推導(dǎo)的結(jié)論是一致的。采用體外DDI和臨床數(shù)據(jù)構(gòu)建的PBPK模型再次確認了MMAE的低CYP3A抑制風(fēng)險�。然而,毒素分子作為DDI victim的風(fēng)險是存在的�,即毒素有可能是肝藥酶或轉(zhuǎn)運體的底物,與誘導(dǎo)或抑制酶�、轉(zhuǎn)運體的藥物聯(lián)用�,可能會影響ADC產(chǎn)品的療效或安全性�。比如MMAE、DM1�、DXd均是CYP3A的底物。
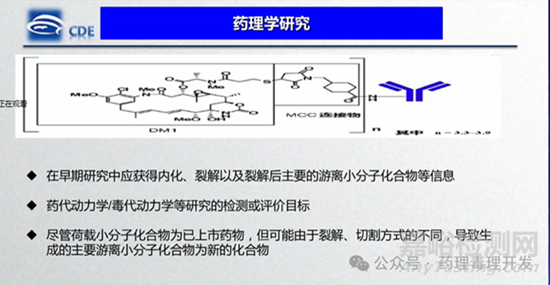
穩(wěn)定性研究:連接子的穩(wěn)定性是影響ADC安全性最重要的因素之一�。這一觀點在ICH S6(R1)和S9指南中均有所體現(xiàn)�,ADC的穩(wěn)定性應(yīng)在人以及非臨床種屬的血漿中進行評估。盡管尚未建立評估ADC穩(wěn)定性的科學(xué)最佳實踐方法�,但體外穩(wěn)定性通常通過在37℃下,持續(xù)3到7天�,測定人以及非臨床種屬血漿中藥物抗體比(DAR)的變化。從監(jiān)管角度來看�,這些研究的主要目的是確定重復(fù)給藥毒理學(xué)研究獲得的數(shù)據(jù)是否能夠代表潛在的人體情況。另外�,在進入IND enabling之前的早期研究中,就應(yīng)獲得ADC內(nèi)化�、裂解及裂解后主要游離小分子化合物信息,這點CDE之前的培訓(xùn)中也有提示�,如下圖所示。血漿穩(wěn)定性研究是小分子體外ADME中的常規(guī)內(nèi)容之一�。
ICH S9指南還要求,關(guān)鍵毒性研究中的毒代動力學(xué)評估(需關(guān)聯(lián)毒性發(fā)生情況的動物PK數(shù)據(jù))需同時涵蓋完整ADC和游離載荷�。常用方法為檢測給藥后血清或血漿中ADC、總抗體(ADC加未結(jié)合抗體)及游離載荷的濃度�,并計算各分析物的PK參數(shù)�。
ADC需驗證多種不同的生物分析方法�。血清中ADC和總抗體濃度通常采用配體結(jié)合試驗檢測,血漿中游離載荷則通過液相色譜-質(zhì)譜聯(lián)用技術(shù)定量�。
毒理學(xué)研究
種屬選擇
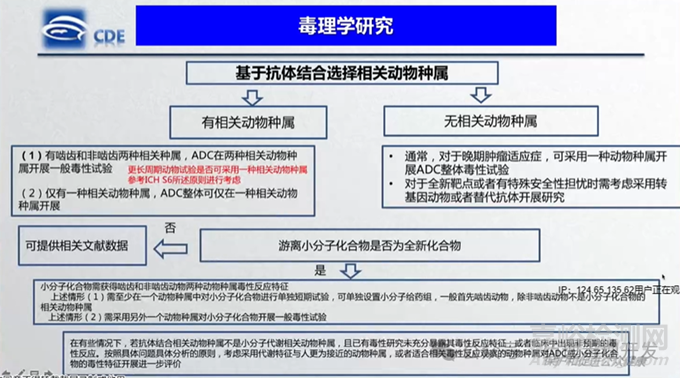
ADC毒理研究的種屬選擇遵循與單抗相同的原則。通常需要評估兩個種屬�,一種嚙齒類動物和一種非嚙齒類動物。當(dāng)然�,必須選擇具備藥理學(xué)活性的相關(guān)動物種屬進行評估。ICH S9 Q&A進一步指出�,當(dāng)ADC的抗體部分僅與人和非人靈長類動物(NHP)抗原結(jié)合時,僅在NHP中進行安全性評估是合適的�。事實上,與大多數(shù)治療性單抗一樣�,ADC與靶點之間的高親和力相互作用通常會排除嚙齒類動物的交叉反應(yīng)性。
那么如何確定相關(guān)動物種屬呢�?首先,應(yīng)基于已發(fā)表的氨基酸序列數(shù)據(jù)�,比較人類與常用毒理研究種屬(如小鼠、大鼠�、犬、小型豬和NHP)之間的靶點序列同源性�。同源性越低,種屬的相關(guān)性越小�。此外,可進一步評估ADC與不同毒理研究種屬和人體之間的靶點結(jié)合親和力�,可通過表面等離子體共振分析或細胞結(jié)合實驗完成�。如果某個種屬的靶點親和力較低�,則可能無法檢測到靶點相關(guān)的毒性。如果與人的結(jié)合親和力相差超過10倍�,可能需要在確定首次人體(FIH)劑量水平時增加額外的安全系數(shù)。當(dāng)然�,親和力還只是藥理作用發(fā)揮的前提,如果條件允許�,最好有功能相關(guān)的數(shù)據(jù)支持。另外�,通過免疫組化或RNA表達譜可以探索靶點表達和分布�,識別潛在的靶器官,并判斷人類與毒理研究種屬之間是否存在相似性(靶向)或差異(非靶向)�。
如果沒有相關(guān)動物種屬怎么辦呢?根據(jù)ICH S6建議�,當(dāng)無法找到相關(guān)動物種屬(即受試物無法與任何動物種屬中的同源靶點相互作用)時,可以考慮使用替代分子或轉(zhuǎn)基因模型�。以Polatuzumab vedotin為例,一款靶向CD79b的ADC�,CD79b表達于B細胞。盡管人和食蟹猴的CD79b表位之間僅存在三個氨基酸的差異�,但該ADC僅能結(jié)合人體靶點,即不存在相關(guān)動物種屬�。因此,開發(fā)了一種能夠結(jié)合食蟹猴CD79b且包含相同連接子-載荷的替代ADC�,用于安全性評估�。在進行毒理研究之前�,比較了替代ADC和人類ADC的活性。
Polatuzumab vedotin和替代ADC分別對人類和食蟹猴的B細胞具有相似的結(jié)合親和力�。此外,體外血漿穩(wěn)定性�、體內(nèi)抗腫瘤活性以及小鼠藥代動力學(xué)數(shù)據(jù)也顯示了兩種ADC之間的可比性。盡管這不是監(jiān)管要求�,但Polatuzumab vedotin和替代ADC均在食蟹猴的GLP毒性研究中進行了給藥。正如預(yù)期的那樣�,只有接受替代ADC的動物外周CD20陽性B細胞數(shù)量減少。然而�,替代ADC和Polatuzumab vedotin均在食蟹猴中引起了載荷相關(guān)的毒性。
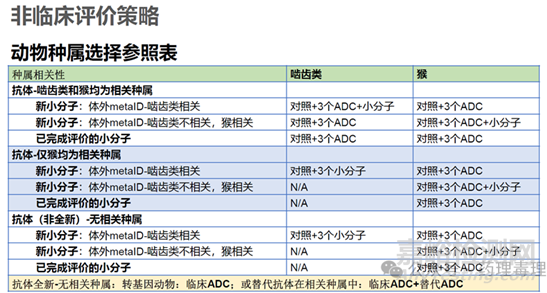
ADC結(jié)構(gòu)中畢竟含有小分子載荷組分�,所以還要額外考慮載荷的安全性評估。對于載荷�,種屬選擇標(biāo)準(zhǔn)則要遵循小分子路徑。不過�,畢竟載荷本身并非臨床試驗藥物,因此可以僅做有限性評估�。對于已有充分研究信息的載荷,比如MMAE�、MMAF、DM-1�、Dxd等等,載荷毒性可以通過整體ADC的給藥實現(xiàn)�。對于新分子或毒性特征不明確的載荷�,根據(jù)NMPA《抗體偶聯(lián)藥物非臨床研究技術(shù)指導(dǎo)原則》要求�,需要按照創(chuàng)新小分子化合物的一般毒性研究原則,獲得其在嚙齒類和非嚙齒類動物中的毒性反應(yīng)信息�,且需要至少在一個動物種屬中進行單獨的毒性考察,該考察可以是采用游離小分子化合物給藥的獨立試驗�,也可以在 ADC 的毒性研究中設(shè)置游離小分子化合物給藥組。根據(jù)抗體和載荷的相關(guān)種屬情況�,會有幾種不同的策略,之前益諾思分享過一個表格�,比較直觀,此處借用�、分享如下。簡單一句話就是�,對于載荷來講�,嚙齒類相關(guān),則首選嚙齒�,反之,僅猴相關(guān)�,則選猴。大部分情況應(yīng)該是�,抗體部分僅猴為相關(guān)種屬,載荷部分則大鼠和猴均相關(guān)�,故開展ADC猴毒理+單獨載荷大鼠毒理組合研究?;蛘呖贵w部分和載荷部分大鼠和猴均相關(guān)�,則開展ADC大鼠和猴毒理�,同時大鼠試驗中增設(shè)1組單獨的載荷組。
關(guān)于這部分�,CDE也分享過資料,如下圖所示�,邏輯是一樣的。
支持IND的一般毒理研究
ICH S9 明確指出�,對于結(jié)合型產(chǎn)品,完整ADC的安全性是主要關(guān)注點�。在S9問答文件中進一步明確,ADC的整個分子應(yīng)在至少一個種屬中進行測試�,單抗不必單獨研究。此外�,對于未結(jié)合的物質(zhì)(如載荷)的安全性評估,可以進行有限的評估�。關(guān)于載荷的毒性評價,ICH S6建議遵循新化學(xué)實體的方法�,即進行完整的包括兩個種屬的非臨床毒理學(xué)評估。當(dāng)然�,抗腫瘤產(chǎn)品還是以ICH S9要求為主。
在GLP非臨床研究中�,藥物給藥途徑必須與臨床試驗中使用的途徑完全一致。生物制劑容易降解�,因此給藥途徑通常限于注射方式,如靜脈輸注、皮下注射�、皮內(nèi)注射或肌肉注射。如果需要改變臨床給藥途徑�,則需要額外的動物研究來評估藥物分布的變化并確定安全性。
初步安全性評估可能包括劑量范圍探索研究或其他非GLP研究�,以幫助確定最大耐受劑量(MTD)并選擇用于正式GLP研究的劑量水平。對于ADC分子�,不需要進行急性毒性研究。重復(fù)給藥毒性研究包括對動物健康狀況的評估�,包括行為變化、外觀�、體重、攝食量�、臨床病理學(xué)參數(shù)以及器官和組織的顯微鏡檢查等。重復(fù)給藥毒性試驗中應(yīng)考慮藥物毒性靶器官�,并在適當(dāng)?shù)那闆r下評估潛在的可逆性。通過這些研究�,可以為FIH臨床試驗的起始劑量選擇提供依據(jù)。
支持IND的GLP研究的給藥周期應(yīng)支持預(yù)期的臨床給藥計劃�,通常取決于分子的半衰期。單抗的半衰期通常支持每周或更少頻率的給藥�。在非臨床和臨床研究中�,ADC的給藥頻率傾向于每3周給藥一次(Q3W),以提高對載荷的耐受性�。ADC在第1周給藥,隨后2周不給藥,視為一個治療周期�。根據(jù)這種給藥方案,單次給藥后進行2周觀察期通常被認為可以支持FIH試驗�。然而,S9問答文件特別指出�,毒性研究中至少需要兩次ADC給藥以支持初始臨床試驗(即在研究第1天和第22天給藥)。需要注意的是�,第二次給藥后的毒性通常大于第一次給藥后的毒性。動物毒性研究中的給藥頻率應(yīng)支持臨床中的給藥頻率�,因此毒性研究的給藥間隔可以設(shè)為2周或3周。
以曲妥珠單抗美坦新(trastuzumab emtansine�,T-DM1)為例,進行了兩項重復(fù)給藥的非人靈長類動物(NHP)研究�。第一項研究為4次重復(fù)給藥,每3周給藥一次(Q3W)�,劑量范圍為3~30 mg/kg,最長恢復(fù)期為6周�。TCR研究確認了人和NHP正常上皮組織中HER2的表達情況。載荷DM1的作用機制是抑制微管聚合�,導(dǎo)致有絲分裂中期阻滯。T-DM1相關(guān)發(fā)現(xiàn)包括肝臟�、骨髓/血液學(xué)(主要是血小板)、淋巴器官毒性以及處于有絲分裂中期阻滯的上皮細胞和髓系細胞數(shù)量增加�。
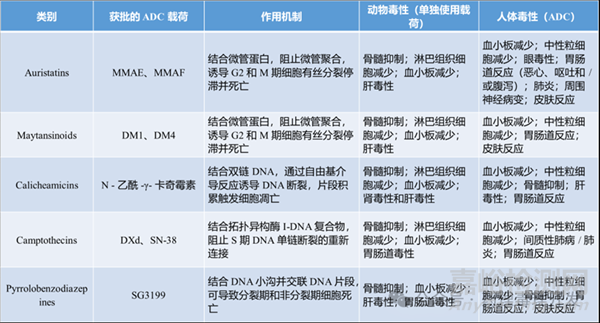
ADC中的載荷部分通常是一種毒素,用于殺死ADC靶向的癌細胞�。盡管ADC的安全性評估是最重要的�,但動物中觀察到的毒性通常與載荷有關(guān)�。不同的載荷類別對應(yīng)的特定毒性已大致清楚。如果載荷之前已經(jīng)經(jīng)過評估�,則可能不需要額外進行GLP毒性研究或進一步評估。對于一種新型毒素�,通常需要在毒性研究中進行表征,研究通常在嚙齒動物中進行�。這可能是一項多劑量的單次給藥研究,并隨后進行短暫的恢復(fù)期觀察�。研究需設(shè)置標(biāo)準(zhǔn)的毒理學(xué)參數(shù)(如臨床觀察以及臨床和解剖病理學(xué))。已上市ADC載荷的分類�、作用機制、動物和人體毒性特點總結(jié)如下表所示�。
ADC的最后一個組成部分是連接子,它負責(zé)將毒性載荷與單抗結(jié)合�。單獨研究游離連接子相關(guān)的毒性可能并不必要。在一些毒理學(xué)研究中�,分別對載荷和連接子-載荷進行了考察,結(jié)果顯示毒性相似且與小分子相關(guān)�,這表明與載荷相關(guān)的毒性相比,連接子相關(guān)的毒性較小�。
支持晚期臨床或上市的重復(fù)給藥毒理研究
如果計劃在臨床試驗中改變給藥方案或給藥方式,且與之前在動物研究中考察的不同�,則可能需要額外進行毒性研究。
如果方案不變�,在關(guān)鍵的Ⅲ期臨床試驗之前,可能需要進行一項更長周期的GLP重復(fù)給藥毒性研究�。對于用于腫瘤治療的ADC,通常3個月的研究足以支持后續(xù)開發(fā)�。即使抗癌藥物被證明能延長患者生存期,或應(yīng)用于非立即危及生命的其他腫瘤適應(yīng)證�,通常也不需要額外進行一般毒理學(xué)研究。在目標(biāo)人群中獲得的臨床安全性數(shù)據(jù)比在額外動物研究中生成的數(shù)據(jù)更能用于評估人體風(fēng)險�。
遺傳毒性
生物制品無需進行遺傳毒性研究,因為它們不會與DNA或其他染色體物質(zhì)直接相互作用�。因此,ADC整體無需進行遺傳毒性評估�。然而,ADC的另外兩個組分——連接子和載荷�,則要考慮。
先看下連接子�。連接子分為可切割和不可切割兩類??汕懈钸B接子可在細胞內(nèi)的酶或化學(xué)條件下釋放載荷,而不可切割連接子則始終與抗體共價結(jié)合�。通常無需單獨對連接子進行評估,但進行體外QSAR測試是一個很好的初步判斷方法�。QSAR評估對細菌回復(fù)突變試驗(Ames試驗)的結(jié)果具有很高的預(yù)測性,如果連接子分子在適當(dāng)?shù)腝SAR評估中未觸發(fā)任何潛在致突變結(jié)構(gòu)警報�,則可被視為非致突變性物質(zhì),無需進一步測試�。如果識別出結(jié)構(gòu)致突變性警報�,則需要進一步測試�。載荷也可以包括在QSAR初步評估中。
載荷部分需要進行遺傳毒性評估�。標(biāo)準(zhǔn)的試驗組合包括體外和體內(nèi)試驗,以確定藥物是否誘導(dǎo)遺傳損傷�。通常,體外Ames試驗可能足以支持IND申請�。如果結(jié)果為陽性,則無需進一步測試�。如果Ames試驗為陰性,但體外染色體損傷試驗(如染色體畸變�、微核或小鼠淋巴瘤tk?/?試驗)為陽性,則應(yīng)考慮進行體內(nèi)遺傳毒性測試�。需要注意的是,由于Ames試驗使用的是細菌�,因此僅對真核細胞有毒性的載荷可能會測試為陰性。在啟動2期臨床試驗之前�,需要完成完整的遺傳毒性試驗組合,通常包括兩項額外的研究:體外哺乳動物細胞染色體畸變試驗和體內(nèi)嚙齒動物骨髓微核試驗�。
如果載荷之前已經(jīng)進行過遺傳毒性評估,這些數(shù)據(jù)可能足以支持ADC的后續(xù)開發(fā)�。要根據(jù)載荷具體情況case by case分析。
生殖毒性
根據(jù)ICH S9�,擬用于晚期腫瘤患者的藥物臨床試驗或上市,生育力�、早期胚胎發(fā)育毒性試驗和圍產(chǎn)期毒性試驗不是必需的�。生育力損傷評估可以在一般毒性研究中伴隨考察�。雖不是必需�,抗腫瘤藥物的胚胎-胎兒發(fā)育毒性研究可以在提交上市申請時完成。對于小分子藥物�,應(yīng)在兩個種屬中進行評估;對于生物制品�,應(yīng)在相關(guān)種屬中進行評估。對于具有遺傳毒性和靶向快速分裂細胞的藥物�,這些研究也不需要。ADC的載荷大多屬于這類�,直接靶向微管或DNA來殺死癌細胞,最終破壞細胞分裂�,故大多無需提交。
如果生物制品的目標(biāo)患者群體預(yù)期壽命較長�,則可以考慮在NHP中開展增強的圍產(chǎn)期發(fā)育研究。然而�,在進行此類研究之前,應(yīng)首先考慮通過作用機制或敲除小鼠模型等信息進行生殖風(fēng)險的證據(jù)權(quán)重(WOE)評估�,以評估對胚胎-胎兒發(fā)育的生殖危害。如果WOE明確表明存在風(fēng)險�,則無需進行NHP研究。靶向相同通路的其它藥物的結(jié)果�,也可作為WOE評估證據(jù)之一。例如�,T-DM1的載荷DM1已在小鼠中顯示出胚胎毒性�、致畸性和染色體斷裂性�。大鼠靜脈注射DM1(0.07-0.2 mg/kg)后,其體內(nèi)的血漿濃度與接受3.6 mg/kg T-DM1每三周一次治療的患者體內(nèi)血漿濃度相當(dāng)�。因此,患者將暴露于會導(dǎo)致胎兒損害的DM1濃度�。因此,T-DM1的藥物標(biāo)簽上有黑框警告�,提示胚胎-胎兒毒性。
致癌性
致癌性研究的要求通常取決于藥物類別和適應(yīng)癥�。對于治療晚期癌癥的藥物,不需要進行致癌性研究�。然而,如果ADC用于治療治愈率高且疾病復(fù)發(fā)率低或復(fù)發(fā)延遲時間長的適應(yīng)癥�,則可能需要開展相關(guān)研究。
其他毒性
局部刺激性試驗可考慮伴隨重復(fù)給藥毒理研究開展�。
溶血性研究參見《藥物刺激性、過敏性和溶血性研究技術(shù)指導(dǎo)原則》�。
過敏試驗通常無需開展。
光安全性:在1期臨床試驗前�,應(yīng)評估潛在風(fēng)險,并進行臨床風(fēng)險控制�。應(yīng)在3期前提供符合ICH S10所述原則的光安全性評估。
免疫原性/免疫毒性:免疫原性可伴隨重復(fù)給藥毒性開展�。免疫毒性方面,由于ADC的細胞毒性載荷會導(dǎo)致快速分裂細胞(如骨髓祖細胞)的死亡,免疫抑制是可以預(yù)期的�。對于抗癌藥物而言,標(biāo)準(zhǔn)毒性研究中對免疫細胞(如淋巴細胞和B細胞)的影響評估通常是足夠的�。然而,對于其他適應(yīng)證�,可能需要根據(jù)ICH S8指南中概述的策略視情況進行額外評估。
最后
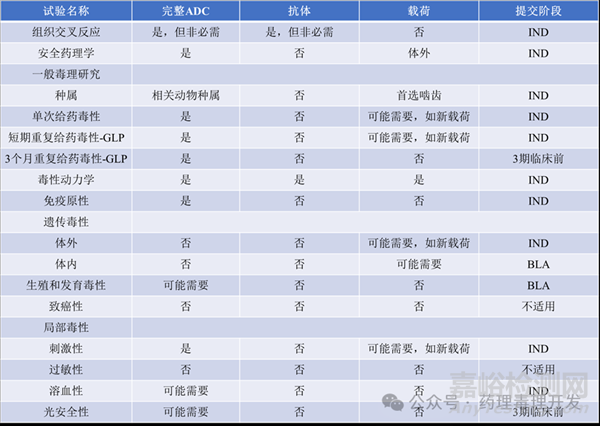
最后大體總結(jié)下非臨床毒理研究需要開展的內(nèi)容�,按照完整ADC、抗體和載荷進行分類�,并羅列了各項研究擬提交的階段�。