在過去十年中����,抗體藥物偶聯物(ADCs)的發(fā)展為提高細胞毒性藥物的療效和降低毒性帶來了希望,與傳統(tǒng)細胞毒性化療藥物相比具有顯著臨床優(yōu)勢�。ADCs將單克隆抗體的精準靶向性、特異性和細胞毒性化療藥物的強大殺傷力相結合���,其核心理念是通過最小化系統(tǒng)毒性來拓寬藥物的治療指數����。一般來說����,ADC的結構組成包括一個靶向腫瘤特異性抗原或相關抗原的抗體���,以及通過連接子連接到抗體上的多個細胞毒性分子(也稱為載荷)。
當ADC進入血液循環(huán)后�,它會選擇性地結合到腫瘤細胞表面表達的受體上���。隨后����,受體-ADC復合物通過內吞作用被內化���。在腫瘤中�,連接子被切割�,細胞毒性載荷被釋放,游離的載荷通過其作用機制誘導細胞死亡���。很多人認為���,ADC設計的關鍵因素主要集中在抗體的選擇上,因為抗體是將細胞毒性藥物遞送至腫瘤的載體�。
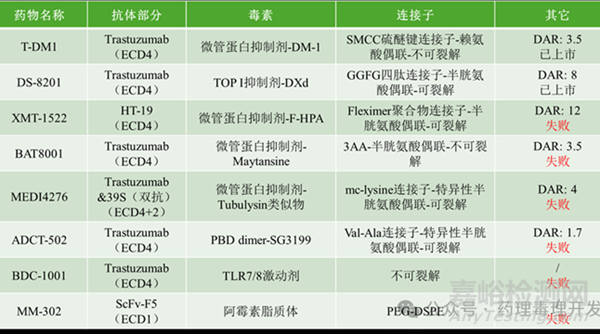
有趣的是���,盡管抗體的選擇很重要�,但ADC的連接子穩(wěn)定性以及載荷的選擇可能對臨床成功更為關鍵����。一個很好的例子是用于治療HER2陽性轉移性乳腺癌的兩種ADC藥物:德曲妥珠單抗(ENHERTU)和恩美曲妥珠單抗(KADCYLA)。在一項三期研究中����,研究者比較了德曲妥珠單抗與恩美曲妥珠單抗在既往接受過曲妥珠單抗和紫杉類治療的HER2陽性轉移性乳腺癌患者中的療效����,主要終點是無進展生存期����。盡管兩種ADC藥物使用了相同的抗體���,但接受德曲妥珠單抗治療的患者疾病進展或死亡的風險顯著低于接受恩美曲妥珠單抗治療的患者�。在524名隨機分配的患者中,12個月時無疾病進展且存活的患者比例在德曲妥珠單抗組為75.8%����,而在恩美曲妥珠單抗組為34.1%���。兩種ADC采用的同一抗體���,差異主要在連接子和載荷方面(如下表示例所示)。
這些結果突顯了在ADC開發(fā)早期階段選擇最合適的載荷分子(以及連接子設計)的重要性。這一理念在當前ADC競爭激烈的環(huán)境中尤為重要。許多ADC藥物在臨床開發(fā)過程中因毒性過大而失敗,包括血液學����、肝臟、神經學和眼部事件等�,這些毒性削弱了藥物的獲益-風險比�。實際上����,許多已獲批的ADC藥物仍有一定比例的患者因ADC相關毒性需要減少劑量����、暫停治療或停止治療。
本文圍繞ADC設計中細胞毒性載荷發(fā)現和開發(fā)的關鍵屬性和考量因素展開����。
常見載荷
ADC本質上是通過攜帶的載荷殺傷癌細胞,載荷的選擇就顯得尤為重要�。在ADC藥物發(fā)展的早期階段���,載荷以傳統(tǒng)的化療藥物如甲氨蝶呤、長春堿和阿霉素等細胞毒性藥物為主����。然而,由于這類ADC藥物對癌細胞的細胞毒性效力不足以及在腫瘤細胞內的蓄積量低����,導致療效一般���,臨床試驗陸續(xù)失敗。研究顯示�,經靜脈給藥后,只有大約1%到2%的ADC分子能夠到達目標腫瘤部位�。這就要求載荷需要具有很高的活性(IC50值處于低納摩爾和皮摩爾水平)����。
目前�,已獲批ADC藥物的載荷主要有兩大類高活細胞毒性藥物:抗微管藥物(例如奧瑞他汀類和美登素類)以及造成DNA損傷的藥物(例如喜樹堿類�、卡利霉素類和吡咯并苯二氮?類)
微管蛋白抑制劑
微管是細胞骨架的主要組成部分�,在細胞分裂過程中發(fā)揮著關鍵作用�,尤其是在腫瘤細胞快速增殖期間����。微管蛋白抑制劑可分為兩類:微管聚合促進劑和微管聚合抑制劑�。這兩類分子均是通過干擾微管依賴的有絲分裂發(fā)揮作用。
奧瑞他?���。ˋuristatin)是一種合成的抗腫瘤藥物,源自天然產物dolastatin 10����。Dolastatin 10本身是一種非特異性毒性藥物,因此不被用作ADC載荷�。然而�,其結構類似物如單甲基奧瑞他汀E(MMAE)通過阻斷微管聚合過程�,導致細胞周期停滯����,已在ADC中大量使用�。例如����,已上市ADC產品Adcetris的細胞毒性載荷就是MMAE�,用于治療復發(fā)或難治性霍奇金淋巴瘤和系統(tǒng)性間變性大細胞淋巴瘤���。
美登素類(Maytansinoids)通過阻止微管二聚體聚合形成成熟微管來抑制有絲分裂和細胞復制����。美登素類是從灌木Maytenus serrata中分離出來的天然產物,目前有兩種美登素衍生物DM1和DM4被用于ADC設計����。其中���,DM1是用于治療HER2擴增型乳腺癌的ADC藥物Kadcyla的彈頭。
DNA損傷劑
顧名思義���,DNA損傷劑是一類能夠對DNA分子造成損傷的化合物���,通過干擾DNA的正常結構和功能,從而抑制細胞增殖、誘導細胞周期停滯或細胞凋亡����。嚴格意義上講,雖然微管破壞劑主要通過干擾微管蛋白的聚合來抑制細胞分裂���,但它們也可以間接導致DNA損傷���,算是DNA損傷劑的一種���。除此以外�,還有兩類主要的DNA損傷劑:一類是拓撲異構酶I抑制劑(Topoisomerase I Inhibitors)����,通過干擾DNA的拓撲結構,阻止DNA復制和轉錄�,從而誘導細胞凋亡����。目前,大多數拓撲異構酶I抑制劑是喜樹堿(camptothecin)的衍生物�,包括拓撲替康(Topotecan)、司他曲安(Govitecan)�、SN-38、Exatecan 和 Deruxtecan 等����。例如����,Enhertu是一種獲批用于治療轉移性HER2陽性乳腺癌的ADC藥物����,載荷用的就是拓撲異構酶I抑制劑�。
另外一類DNA損傷劑是吡咯并苯二氮?類(Pyrrolobenzodiazepines, PBDs),一類從天然產物安曲霉素(anthramycin)衍生而來的DNA損傷劑���。它們通過結合DNA小溝中的5′-嘌呤-鳥嘌呤-嘌呤序列�,并與鳥嘌呤的外環(huán)氨基形成共價鍵,從而干擾DNA的正常功能�。比如Tesirine(SG3199)和 Talirine 等PBDs因其高細胞毒性而被用作ADC的載荷。已上市ADC藥物Zylonta載荷就是PBD�。卡利霉素類(Calicheamicins)也是一類DNA損傷劑����,通過結合DNA小溝���,誘導雙鏈DNA斷裂�,從而導致細胞死亡����。例如,N-乙酰-γ-卡利霉素被用于Mylotarg和Besponsa等ADC藥物的載荷�。
為什么理解載荷的ADME非常重要
ADC的不良反應通常包括靶向作用相關的不良反應和非靶向作用相關的不良反應���,其中非靶向作用相關的不良反應是決定其最大耐受劑量的主要因素����。既然是非靶向作用,這些劑量限制性毒性往往與靶向的抗原和/或治療的癌癥類型無關�,常常限制了ADC的劑量,使其低于能夠實現最佳抗癌效果的劑量����。細胞毒性藥物的低皮摩爾級活性是導致ADC不良反應的重要因素�,因此僅靠細胞毒性藥物的活性���,可能很難實現遠高于1的治療指數。因此����,除了具有極高的活性外,ADC的藥物載荷還必須具備適當的藥物代謝和藥代動力學(DMPK)特性���。
DMPK是藥物發(fā)現過程中的重要組成部分����。在過去十年中����,對藥物在體內的吸收�、分布、代謝和排泄(ADME)以及藥代動力學(PK)特性的理解���,已經從描述性研究轉變?yōu)槎亢蜋C制方面的理解,以了解藥物在體內的過程���。對于傳統(tǒng)的口服小分子藥物而言���,DMPK科學家主要關注兩個方面:化合物是否具有生物藥劑學上合適的����、可成藥的藥代動力學特性�,以及化合物是否具有可能在臨床中引發(fā)安全問題的特性。為此�,清除率和生物利用度是前者的關鍵特征,而藥物相互作用(DDIs)和代謝相關的特異性藥物反應(生物活化)則是后者的首要考慮因素���。在這種背景下����,可以嘗試將ADC藥物載荷的開發(fā)重點與用于口服給藥的小分子藥物進行對比���。
載荷的溶解性
對于口服小分子藥物候選物需具有可接受的生物利用度����。因此,在藥物發(fā)現過程中����,藥物候選物在水性介質中的溶解性是需要優(yōu)化的關鍵理化性質之一�。一般來說,候選藥物需要具有高于10μM的溶解度�,才能進行臨床前測試�。此外,低溶解度分子的口服生物利用度通常變異比較大����,且往往劑量/暴露量之間不成比例,通常需要進行制劑配方優(yōu)化����。
不過,ADC畢竟是靜脈給藥����,看似不涉及口服生物利用度這一問題����。盡管口服吸收與ADC藥物載荷無關����,但仍需了解候選載荷的幾個基本特性:大小���、極性����、親脂性和構象動力學�。這些理化性質很重要�,因為藥物載荷需要具有適合大規(guī)模偶聯到抗體的功能基團���。比如許多高活性小分子藥物�,缺乏所需的化學功能基團用于偶聯����,如果這類載荷進行修改,引入這些“反應基團”����,可能會對藥物載荷的活性產生不利影響���。此外,除了需要具備“反應基團”外����,候選載荷還必須具有足夠的溶解性,因為抗體偶聯過程是在水性環(huán)境中進行的����。為什么不在有機溶劑中呢���?因為����,偶聯反應中過度暴露于有機溶劑可能會使抗體變性,導致蛋白質構象變化和聚集����,從而導致ADC功能喪失,并可能增加免疫原性風險����。
載荷滲透性和旁觀者效應
一般來說�,小分子藥物穿過脂質生物膜的能力是決定其ADME屬性的關鍵����,影響生物利用度(如穿過胃腸道上皮)或藥理學特性(如穿透器官屏障),決定藥物能夠發(fā)揮預期的治療效果���。因此�,藥物的親脂性(描述化合物在水相和脂相之間分配的指標)是藥物發(fā)現過程中需考察的基本理化性質指標。對于口服給藥的分子來說�,滲透性是一個關鍵的指標,與分子口服生物利用度高低直接有關�。盡管口服生物利用度對于ADC的藥物載荷來說并不是一個重要考慮因素,但高滲透性這一特性仍然需要關注�。因為,一些藥物載荷在被內吞并殺傷腫瘤細胞后����,會從最初的腫瘤細胞中逸出�,并穿過鄰近腫瘤細胞的細胞膜,發(fā)揮“旁觀者效應”�,形成不依賴TAA的腫瘤殺傷。這種殺傷效果的強弱對載荷的細胞膜滲透性就有比較高的要求�。李等人(Li and others)研究了藥物載荷旁觀者效應對ADC療效的藥理學作用����,比較了兩種ADC的體內活性���,這兩種ADC具有相同的抗體和連接子���,但藥物載荷不同(MMAE和MMAF)�。在這項研究中���,滲透性更好的MMAE展示了對鄰近腫瘤細胞的強烈旁觀者殺傷作用����,而膜滲透性較差的藥物載荷MMAF未能在體內介導旁觀者殺傷�。
載荷的代謝
另一個支持ADC載荷選擇的關鍵指標之一是未結合藥物的清除速率。一般來說���,藥物在進入體內后�,需要到達作用部位才能發(fā)揮其藥理學效應���。然而����,如果藥物的PK特性不理想(如高清除率和/或短半衰期),那么該分子可能無法實現預期的藥效學(PD)效應���。一般來說�,大多數脂溶性藥物首先在肝臟中被代謝�,然后才能被排泄����。藥物的代謝可以分為I相(修飾)和II相(結合)。I相反應通過氧化�、還原����、水解等方式改變脂溶性藥物的化學結構����,使其成為更具極性的分子�,通常是通過移除氫原子或添加氧原子來實現�。細胞色素P450酶家族負責這些I相反應的大部分過程。利用人和動物肝臟進行的體外代謝研究���,以及在臨床前種屬中進行的體內代謝研究���,是識別藥物代謝途徑的主要方法。通常認為����,苯甲基C-H鍵���、烯丙基甲基和O-�、N-���、S-甲基基團是P450介導的藥物氧化反應的敏感代謝位點���。對于II相代謝����,藥物通過結合反應與另一分子結合�,產生一種藥理學上無活性且有一定水溶性的化合物,從而更容易排泄�。結合機制包括甲基化����、乙?;⒘蛩峄?��、葡萄糖醛酸化以及甘氨酸或谷胱甘肽結合。
小分子藥物代謝的原理與ADC載荷的代謝完全相同���。不過,從ADC釋放的載荷不會像口服小分子藥物那樣經歷首過消除。當然����,ADC載荷會被藥物代謝酶處理,并在未結合載荷消除過程中發(fā)揮重要作用����。因此���,除了了解載荷發(fā)生了哪些化學修飾外���,還需要確定哪些藥物代謝酶促進了載荷的消除。在載荷選擇過程中����,進行肝藥酶表型研究可以幫助識別參與載荷代謝的特定酶���。這些表型研究的結果可以支持后續(xù)的DDI研究�。而且,ADC細胞毒性載荷的劑量反應關系陡峭和治療指數狹窄�,這一試驗結果就顯得尤為重要。畢竟���,多藥聯用以及老年腫瘤患者合并其它用藥���,潛在的DDI風險���,可能會導致載荷系統(tǒng)暴露量的變化���,并可能在某些個體中導致嚴重甚至危及生命的毒性����。比如ADC載荷SN-38���,其常見的嚴重毒性包括中性粒細胞減少癥和腹瀉���。在人體中�,SN-38的消除主要通過UDP葡萄糖醛酸轉移酶(UGT1A1)將藥物結合成β-葡萄糖醛酸苷結合物SN-38G來實現。在臨床中���,由于UGT1A1個體間基因多態(tài)性���,SN-38的葡萄糖醛酸化程度存在很大差異���,其中具有UGT1A1*28多態(tài)性的患者對SN-38的葡萄糖醛酸化能力降低,從而導致更高的SN-38暴露量�。另外,UGT活性的降低與伊立替康(SN-38的前藥)觀察到的毒性風險增加有關�。
雖然載荷的肝臟代謝表征比較重要,但成功的ADC載荷還要關注在腫瘤中的穩(wěn)定性����。畢竟,載荷毒素的藥理學靶點在腫瘤細胞內,因此載荷在釋放到細胞內后必須能夠抵抗降解及隨后的失活�,以保持生化環(huán)境中的穩(wěn)定。例如�,酸敏感載荷在溶酶體中不穩(wěn)定,而含有二硫鍵���、烯烴或環(huán)氧化物等功能基團的載荷可能被酶還原或轉化����,從而影響載荷向其使命終點的遞送���。ADC的體內生命周期通常是這樣的���,首先被靜脈注射進入系統(tǒng)循環(huán),之后識別腫瘤細胞表面表達的目標抗原����,并隨后形成抗原-ADC復合物。接下來����,整個抗原-ADC復合物通過形成有被小窩并進一步發(fā)展為早期內體,內化進入細胞內�。之后,一部分ADC與FcRn結合���,并被反向轉運回到細胞外�。剩余的抗原-ADC復合物經歷早期內體成熟為晚期內體���,之后與溶酶體融合�,并將ADC運輸到降解部位����,從而釋放載荷���。溶酶體通過H+ -ATPase持續(xù)輸入質子,維持pH值在4.5到5.0之間的酸性環(huán)境���,并且還含有高濃度的蛋白水解酶���,如組織蛋白酶和膠原酶。釋放或游離的載荷必須成功地通過內體/溶酶體的考驗���,才能發(fā)揮其預期的藥理學效應。
載荷的未來方向
ADC在實體瘤和血液瘤的全面開花����,吸引了工業(yè)界和學術界的廣泛關注,并開始設計下一代ADC���。下一代ADC主要圍繞三個核心組成部分展開���,即抗體�、連接子和載荷。載荷方面����,兩類非細胞毒性治療藥物——蛋白降解靶向嵌合體(PROTACs)和小干擾核糖核酸(siRNAs)——似乎是理想的ADC載荷����。
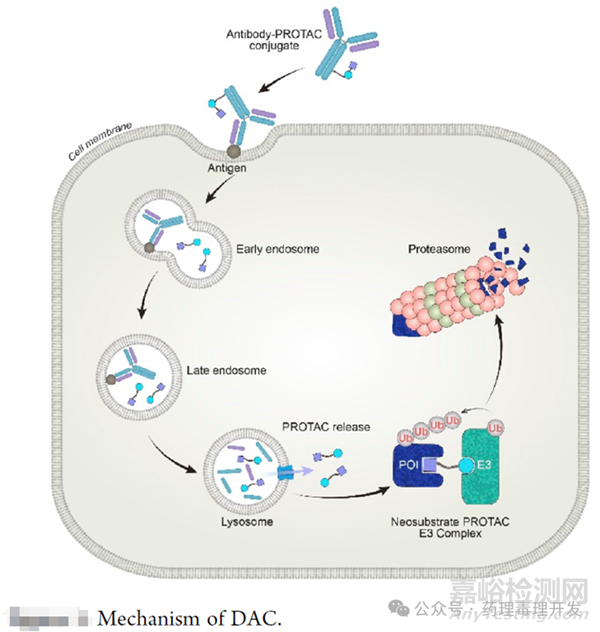
PROTACs是由針對目標蛋白(POI)的配體部分通過化學連接子與E3泛素連接酶配體連接而成的雙功能化合物���。這些分子的藥理作用機制依賴于它們將POI和E3連接酶拉近到足夠近的距離,從而觸發(fā)POI的定向多泛素化和隨后的蛋白酶體介導的降解�。盡管PROTACs是高效的降解劑���,但通常不具備組織特異性�。因此����,通過安裝一種增強組織特異性PROTAC降解的手段(例如ADC),可以減少PROTACs的副作用�,優(yōu)化臨床治療窗口���,從而增強其在癌癥治療中的潛力。當然,抗體與分子膠偶聯也能達到類似目的�。這個領域已經產生了不少的交易���。2023年9月7日����,Seagen與Nurix簽訂一項合作協議�,共同開發(fā)抗體偶聯降解劑(Degrader-Antibody Conjugates,DAC)����,Nurix將獲得6000萬美元的預付款����,并有可能獲得高達約34億美元的多個項目的研究、開發(fā)、監(jiān)管和商業(yè)里程碑付款����。2023年11月,Orum公司宣布與BMS簽署了一項協議�,BMS以1億美元預付款收購了Orum的靶向蛋白降解劑ORM-6151,并有望獲得總交易價值約1.8億美元的里程碑付款�。2023年12月12日,C4 Therapeutics宣布與默沙東簽訂協議����,共同開發(fā)DAC���。作為合作的一部分,C4T將負責開發(fā)有效載荷����;默沙東將負責抗體偶聯,在藥物發(fā)現階段創(chuàng)建DAC���。C4T將獲得1000萬美元的預付款�;對于合作的首個靶點,C4T有望獲得總計約6億美元的里程碑付款�,以及未來銷售的分級特許權使用費。此外�,默沙東還擁有三個合作項目的擴大選擇權����,倘若行使權利���,C4T將有資格在整個合作中獲得高達約25億美元的潛在付款���。
siRNAs是一類屬于小非編碼核糖核酸(sncRNA)家族的藥物����,能夠通過與mRNA的特異性序列識別來靶向并沉默特定基因表達�,例如癌基因�、腫瘤抑制基因和其他調控基因�。siRNAs存在兩點局限性,限制了其成藥性�。一是大小和高負電荷����,使其無法通過被動攝取進入細胞;二是未修飾的siRNAs通常在生理條件下半衰期較短�,因為容易被細胞外和細胞內核酸酶快速降解���。然而�,通過使用選定的連接子和偶聯化學將這類分子與靶向大分子結合�,生成一種新的ADC設計,可能會增強這類分子的成藥性�。
引自:Beyond Cytotoxic Potency: Disposition Features Required to Design ADC Payload