一����、四環(huán)素類(lèi)藥物簡(jiǎn)介
四環(huán)素類(lèi)抗生素是一類(lèi)具有氫化并四苯基本骨架的廣譜抗生素����。20世紀(jì)40年代首個(gè)四環(huán)素類(lèi)藥物—金霉素被發(fā)現(xiàn),迄今為止�����,四環(huán)素類(lèi)藥物已在臨床應(yīng)用中超過(guò)80年�����。盡管耐藥性問(wèn)題日益突出��,但四環(huán)素類(lèi)藥物憑借其獨(dú)特的抗菌譜、抗炎特性及多效性作用����,在現(xiàn)代醫(yī)療中仍占據(jù)著不可替代的重要地位。例如多西環(huán)素和米諾環(huán)素�����。
根據(jù)發(fā)現(xiàn)順序和結(jié)構(gòu)修飾��,四環(huán)素類(lèi)藥物可分為三代�����,第一代為天然抗生素���,主要從土壤放線菌中分離而得,包括金霉素����,土霉素和四環(huán)素。金霉素最早被發(fā)現(xiàn)��,現(xiàn)在主要用于獸藥或眼膏�����。土霉素抗菌活性較弱,除人用外��,大多作為水產(chǎn)養(yǎng)殖用藥物����。四環(huán)素是該類(lèi)藥物的原型,曾廣泛應(yīng)用��,但耐藥性及藥代動(dòng)力學(xué)特性較差��。目前四環(huán)素全身性應(yīng)用已被新一代藥物取代���。
第二代四環(huán)素類(lèi)藥物包括多西環(huán)素和米諾環(huán)素�。多西環(huán)素又名強(qiáng)力霉素�����。其口服吸收好�����,半衰期長(zhǎng)(約18小時(shí))�����,可每日給藥1-2次。由于并四苯骨架極性非常低��,多西環(huán)素脂溶性強(qiáng)�,組織穿透力強(qiáng),尤其在前列腺�、肺組織和泌尿生殖道中濃度高。因其優(yōu)異的安全性譜和藥代動(dòng)力學(xué)特性����,已成為治療上述組織感染癥的首選。米諾環(huán)素脂溶性極強(qiáng)����,組織穿透性極佳�,甚至可穿過(guò)血腦屏障。常用于治療痤瘡和某些神經(jīng)系統(tǒng)感染�����,但前庭毒性相對(duì)較高���,不良反應(yīng)為頭暈����、眩暈。第三代四環(huán)素藥物如替加環(huán)素����。
二、四環(huán)素類(lèi)藥物的配位作用及影響
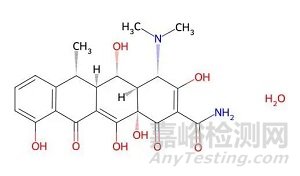
以強(qiáng)力霉素為例�,其配位作用主要發(fā)生于以下兩個(gè)區(qū)域:
1.主配位域,位于A環(huán)C1與C3位點(diǎn)之間的β-二酮系統(tǒng)�。在酸性及中性色譜條件下,該結(jié)構(gòu)可發(fā)生烯醇化�����,形成一個(gè)帶負(fù)電荷的���、高度離域的π電子體系��,構(gòu)成一個(gè)理想的二齒或三齒配體����。
2.輔助配位域��,B環(huán)C10位的酚羥基與C11、C12位的羰基可共同構(gòu)成另一個(gè)潛在的金屬結(jié)合位點(diǎn)����。
圖1強(qiáng)力霉素分子結(jié)構(gòu)圖
四環(huán)素類(lèi)藥物的核心作用機(jī)制是藥物與細(xì)菌核糖體30S亞基的A位點(diǎn)可逆性結(jié)合。具體來(lái)說(shuō)��,藥物與核糖體上Mg2+位點(diǎn)產(chǎn)生配位作用�����,形成空間位阻�����,阻止氨酰-tRNA進(jìn)入A位點(diǎn)�����,從而抑制細(xì)菌蛋白質(zhì)的合成����,產(chǎn)生抑菌效應(yīng)(在高濃度下表現(xiàn)為殺菌作用)�。
四環(huán)素的配位特性是一把雙刃劍,既使其具有抗菌活性�����,又帶來(lái)諸多不利因素。首先四環(huán)素類(lèi)藥物與金屬離子的配位作用��,導(dǎo)致用藥安全性問(wèn)題非常突出�����,口服后四環(huán)素類(lèi)藥物可與胃腸道內(nèi)的二價(jià)(Ca2+, Mg2+)和三價(jià)(Al3+, Fe3+)陽(yáng)離子形成不溶性螯合物�����,嚴(yán)重影響吸收���,產(chǎn)生胃腸道反應(yīng)�,常見(jiàn)惡心�、嘔吐、腹瀉����、食管潰瘍等。四環(huán)素類(lèi)藥物可與發(fā)育中的牙齒和骨骼中的鈣產(chǎn)生配位作用�,導(dǎo)致四環(huán)素牙和骨骼生長(zhǎng)抑制,藥物沉積于牙釉質(zhì)�����,引起牙齒永久性黃染、棕染(熒光性)和釉質(zhì)發(fā)育不良��。藥物沉積于骨骼����,則會(huì)暫時(shí)性抑制骨骼生長(zhǎng)。
四環(huán)素的配位特性也嚴(yán)重影響了其分析檢測(cè)���。目前大多使用液相色譜和液相色譜-串聯(lián)質(zhì)譜方法對(duì)四環(huán)素藥物進(jìn)行分析檢測(cè)����。然而���,在常規(guī)反相色譜條件下����,四環(huán)素類(lèi)化合物常表現(xiàn)出異常的色譜行為�,如嚴(yán)重的峰形拖尾,影響定量的準(zhǔn)確性與可靠性�。
四環(huán)素類(lèi)化合物色譜行為異常���,根源在于其分子對(duì)金屬離子的配位能力?,F(xiàn)代色譜系統(tǒng),為提高柱效�����,正逐步使用細(xì)顆粒色譜柱����,顯著提高了系統(tǒng)的壓力,不銹鋼流路(柱管��、篩板��、管路等)是應(yīng)對(duì)高系統(tǒng)壓力的主要技術(shù)手段����。
當(dāng)四環(huán)素類(lèi)藥物流經(jīng)色譜柱及管路時(shí),其A環(huán)的β-二酮系統(tǒng)作為強(qiáng)Lewis堿����,與系統(tǒng)內(nèi)暴露的酸性金屬離子產(chǎn)生強(qiáng)烈的配位作用,這種相互作用的強(qiáng)度遠(yuǎn)超反相色譜中的疏水分配作用����,且配位平衡動(dòng)力學(xué)過(guò)程緩慢���,導(dǎo)致部分分析物分子被暫時(shí)吸附于色譜柱特定活性位點(diǎn)上,在色譜圖上表現(xiàn)為嚴(yán)重的峰拖尾����。對(duì)于配位作用更強(qiáng)的化合物,甚至發(fā)生不可逆吸附��,導(dǎo)致絕對(duì)回收率下降��。另外配位作用也可能造成保留時(shí)間的重現(xiàn)性差�,從而影響準(zhǔn)確定性。
配位作用是四環(huán)素類(lèi)藥物發(fā)揮抗菌藥理作用(與核糖體30S亞基上Mg²?結(jié)合)的結(jié)構(gòu)基礎(chǔ)����。然而,在分析檢測(cè)場(chǎng)景下����,此特性成為獲得理想色譜數(shù)據(jù)的核心障礙。
三�、如何構(gòu)建惰性色譜分析體系
降低金屬活性表面的影響是解決此問(wèn)題的一個(gè)途徑。因此需要建立一個(gè)涵蓋色譜柱����、流動(dòng)相及整個(gè)流路系統(tǒng)的金屬惰性分析體系�。
3.1 選取金屬鈍化色譜柱
現(xiàn)代反相色譜柱包括填料和柱體兩部分���,填料使用的硅膠顆粒大都采用高純度硅膠合成工藝,極大降低了金屬雜質(zhì)的含量�����,消除了潛在的配位相互作用位點(diǎn)��。但常規(guī)反相色譜柱不會(huì)對(duì)金屬柱體進(jìn)行處理�,因此常常存在活性表面?�?蛇x擇金屬鈍化色譜柱����,此類(lèi)色譜柱對(duì)不銹鋼組件進(jìn)行特殊表面處理,如形成致密的鉻氧化物鈍化層或鍍覆聚合物薄膜�����,構(gòu)成物理屏障�,阻斷分析物與金屬的接觸。
3.2 流動(dòng)相體系的選擇
需選擇合適的流動(dòng)相以抑制次級(jí)相互作用�,同時(shí)兼容質(zhì)譜檢測(cè)��。
水相宜采用含2-10 mM 甲酸銨和0.1%(v/v)甲酸的水溶液�。甲酸提供酸性環(huán)境(pH ≈ 2.5)�,確保硅醇基與分析物上的堿性基團(tuán)質(zhì)子化,最大限度地抑制離子交換相互作用�;甲酸還可提供H+,有助于正離子模式下形成穩(wěn)定的[M+H]?準(zhǔn)分子離子��。甲酸銨提供揮發(fā)性緩沖體系��,穩(wěn)定pH�����,保證保留時(shí)間的重現(xiàn)性����。
有機(jī)相選擇乙腈,因其具有較低的粘度����、較小的離子抑制效應(yīng)及適宜的洗脫強(qiáng)度。
水��,甲酸,甲酸銨和乙腈宜選擇質(zhì)譜級(jí)試劑���,最大程度減少干擾離子����,儲(chǔ)存有機(jī)相的玻璃容器應(yīng)采用的高硼硅玻璃����,以減少金屬離子析出�����。使用PP(聚丙烯)或PET(聚對(duì)苯二甲酸乙二醇酯)材質(zhì)的容器儲(chǔ)存水相流動(dòng)相�����,最大程度避免金屬離子干擾����。
4.實(shí)例:四種四環(huán)素類(lèi)藥物的LC-MS/MS分析
為分析檢測(cè)四環(huán)素,土霉素����,金霉素,強(qiáng)力霉素這四種應(yīng)用廣泛的四環(huán)素類(lèi)藥物��,建立了強(qiáng)力霉素的液相色譜-串聯(lián)質(zhì)譜分析方法。
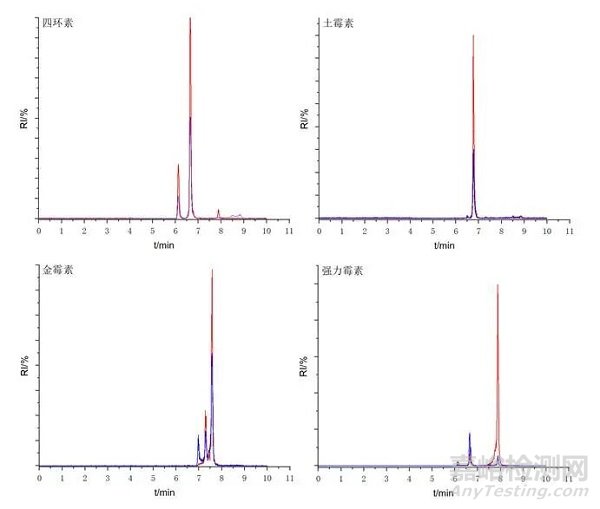
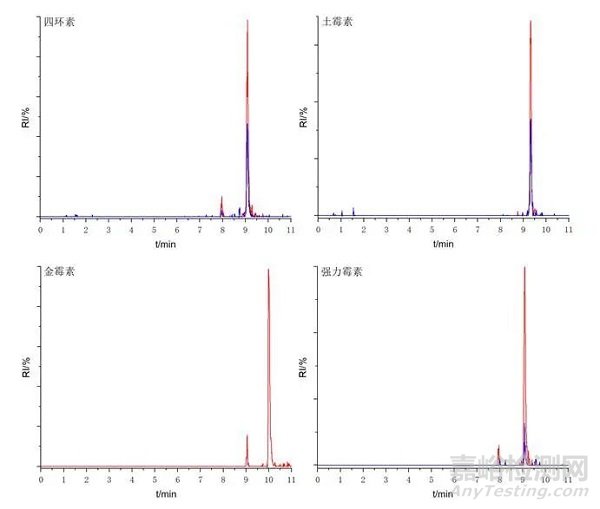
首先采用金屬表面經(jīng)特殊鈍化處理的雜化顆粒C18色譜柱(2.1 × 100 mm, 1.7 μm)�,并與未經(jīng)表面特殊處理的雜化顆粒C18色譜柱(3.0× 100 mm, 2.7 μm)進(jìn)行對(duì)比。
流動(dòng)相水相采用含5 mmol/L 甲酸銨的0.1%甲酸水溶液����,有機(jī)相則使用乙腈,儲(chǔ)存水相的容器采用PET材質(zhì)的塑料瓶�����,有機(jī)相則采用Duran玻璃(一種高硼硅玻璃)材質(zhì)的容器儲(chǔ)存��。
液相系統(tǒng)中����,除高壓區(qū)采用金屬材質(zhì),低壓區(qū)一律更換為耐高壓PEEK(聚醚醚酮)輸液管��,降低金屬對(duì)分析檢測(cè)目標(biāo)物的影響����。
梯度洗脫條件如下:0-1 min,水相比例90%��,有機(jī)相比例10%,清洗樣液中的極性物質(zhì)�;1-9 min,有機(jī)相比例線性增加至90%�,完成化合物分離;9-11 min����,保持90%有機(jī)相比例2min,洗脫待測(cè)化合物�����,并對(duì)色譜柱中極性較低的物質(zhì)進(jìn)行清洗��;11.01-13 min���,將水相比例快速調(diào)整至90%,并保持3 min����,充分平衡色譜柱,保證重現(xiàn)性���。
質(zhì)譜條件��,電噴霧離子源(ESI+)���;毛細(xì)管電壓��,3.5 KV��;RF電壓����,2.5 V�����;萃取電壓����,3 V;源溫���,150°C�����;脫溶劑氣溫度�����,500°C�;脫溶劑氣流速:1000 L/Hr;錐孔氣流速���,50 L/Hr�。
分析方法驗(yàn)證數(shù)據(jù)表明�����,采用該策略后�����,金霉素和強(qiáng)力霉素的色譜行為得到根本性改善��,改善了峰拖尾現(xiàn)象�����,峰對(duì)稱因子達(dá)到0.95-1.05���,四環(huán)素和土霉素的峰寬由0.5 min降至0.25 min����?���?梢宰C實(shí)金屬配位作用得到控制。
圖2 未經(jīng)金屬鈍化處理的C18色譜柱色譜圖
圖3鈍化處理的C18色譜柱色譜圖
連續(xù)進(jìn)樣中�,保留時(shí)間的相對(duì)標(biāo)準(zhǔn)偏差優(yōu)于1.5%。由于峰形對(duì)稱����,積分更加準(zhǔn)確,線性范圍得到顯著改善�。不同濃度水平下的回收率可以滿足定量要求。
5. 總結(jié)
四環(huán)素類(lèi)化合物在色譜分析中的峰形劣化問(wèn)題�,其本質(zhì)可能是分析系統(tǒng)活性位點(diǎn)β-二酮系統(tǒng)與金屬的配位作用。有效的解決方案是采用金屬鈍化色譜柱及惰性流路的液相色譜系統(tǒng)�����。本文實(shí)驗(yàn)部分充分驗(yàn)證了該方案可顯著減輕峰拖尾��,實(shí)現(xiàn)穩(wěn)定準(zhǔn)確的定量�。
可以推測(cè),本文的方法具有普適性����,可推廣至所有易與金屬發(fā)生配位作用的化合物分析�,如核苷酸��、磷酸化肽段����、兒茶酚胺類(lèi)物質(zhì)等。本文方法為易于和金屬配位的化合物分析提供了有益借鑒��。