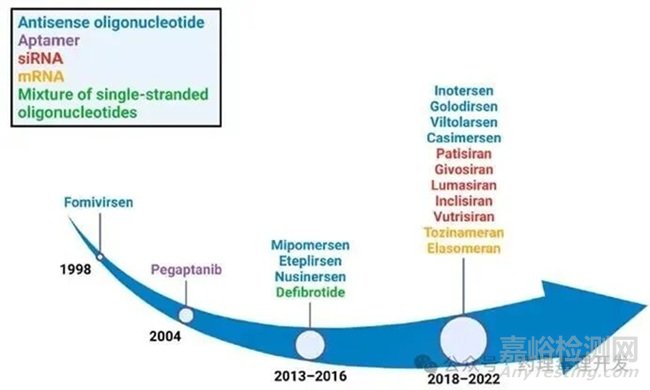
在近幾十年中,RNA治療藥物在罕見(jiàn)病以及抗擊COVID-19疫苗領(lǐng)域取得了很大突破����。RNA治療藥物通過(guò)下調(diào)靶基因調(diào)節(jié)致病蛋白的翻譯����,或者通過(guò)注射合成的mRNA來(lái)翻譯編碼的目標(biāo)蛋白。因此,RNA治療藥物可以被設(shè)計(jì)為靶向幾乎任何遺傳元件��,其中許多在過(guò)去可能被認(rèn)為對(duì)小分子化藥或生物藥來(lái)講不可成藥的靶點(diǎn)。過(guò)去這些年��,F(xiàn)DA已經(jīng)批準(zhǔn)了眾多RNA治療藥物,包括反義寡核苷酸(ASOs)��、小干擾RNA(siRNA)等�。
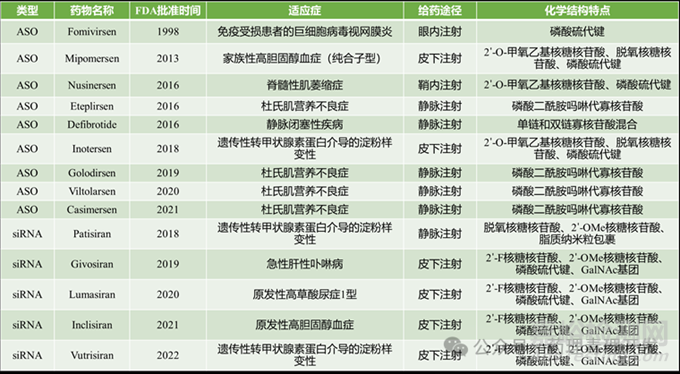
本文對(duì)部分已上市的寡核苷酸藥物(截至2022年)的臨床藥代動(dòng)力學(xué)(PK)特點(diǎn)進(jìn)行總結(jié)。2016年��,Nusinersen被FDA批準(zhǔn)用于治療脊髓性肌萎縮癥����,成為首個(gè)獲批用于該疾病的藥物����。Eteplirsen則是首個(gè)獲批用于治療某些類型杜氏肌營(yíng)養(yǎng)不良癥(DMD)的藥物。在首個(gè)DMD治療藥物獲批之后��,Golodirsen、Viltolarsen和Casimersen分別于2019年�、2020年和2021年被FDA批準(zhǔn)����。此外�,在2018年首個(gè)siRNA藥物Patisiran獲批之后����,每年都有新的siRNA藥物獲批��。已獲批的部分寡核苷酸藥物基本信息列表總結(jié)如下(截至2022年)�。
RNA藥物的結(jié)構(gòu)和堿基修飾
RNA治療藥物是由RNA堿基(腺嘌呤����、胞嘧啶、鳥(niǎo)嘌呤和尿嘧啶)組成的寡核苷酸��。每個(gè)堿基都連接到磷酸骨架上�,為鏈狀結(jié)構(gòu)提供支持����。RNA治療藥物通過(guò)沃森-克里克堿基配對(duì)原則與前體或成熟RNA的特定位點(diǎn)結(jié)合,以發(fā)揮其治療作用�。根據(jù)結(jié)構(gòu)特點(diǎn),RNA治療藥物可以分為ASOs�、siRNA、mRNA和核酸適配體����。由于缺乏傳統(tǒng)藥代動(dòng)力學(xué)研究��,故mRNA疫苗不在討論之列。歷史上����,唯一獲得FDA批準(zhǔn)的基于核酸適配體的藥物是Pegaptanib��。Pegaptanib是一種含有27個(gè)堿基的RNA核酸適配體��,能夠抑制VEGF,用于治療年齡相關(guān)性黃斑變性��。然而��,由于更有效的治療選擇(如雷珠單抗)的出現(xiàn)�,其使用逐漸減少�,最終停止銷售����。
Defibrotide是一種由單鏈和雙鏈寡核苷酸組成的多分散混合物,于2016年首次獲批��,用于治療肝靜脈閉塞性疾病��,可能對(duì)保護(hù)肝臟血管內(nèi)皮細(xì)胞和防止血液凝固起到作用����。Defibrotide具有多種復(fù)雜的作用機(jī)制,包括抗炎�、抗動(dòng)脈粥樣硬化��、抗缺血和抗血栓特性��。
過(guò)去幾年獲批的大多數(shù)RNA治療藥物為ASOs或siRNA藥物。ASOs是長(zhǎng)度為12–25個(gè)核苷酸的互補(bǔ)核酸片段�,被設(shè)計(jì)為特異性地與內(nèi)源性前mRNA或mRNA的互補(bǔ)序列雜交,以調(diào)節(jié)生物功能��。它們通過(guò)以下方式實(shí)現(xiàn)這一目標(biāo):通過(guò)酶促裂解降解mRNA,改變前mRNA剪接以包含或排除特定內(nèi)含子/外顯子����,或者改變調(diào)控RNA的功能�。其他潛在的ASOs作用機(jī)制還包括通過(guò)阻礙核糖體活性誘導(dǎo)翻譯停滯�、通過(guò)抑制剪接干擾前mRNA成熟�、在細(xì)胞核內(nèi)使前mRNA不穩(wěn)定��,或者糾正隱性剪接位點(diǎn)��。siRNA具有明確的結(jié)構(gòu)����,由短的(通常為20-24個(gè)堿基對(duì))RNA雙鏈組成����,3'端有2個(gè)堿基的突出��。siRNA利用人Ago 2蛋白形成RNA誘導(dǎo)沉默復(fù)合物(RISC),進(jìn)而介導(dǎo)靶mRNA的切割和降解����,從而阻止了mRNA的翻譯��,實(shí)現(xiàn)了對(duì)特定基因表達(dá)的沉默。Patisiran是2018年獲批的首個(gè)siRNA藥物����,被制成脂質(zhì)納米粒(LNP)以實(shí)現(xiàn)有效的靶向遞送��。它由一個(gè)21個(gè)堿基的雙鏈siRNA(ALN-18328)組成��,包裹在四種脂質(zhì)輔料中,包括兩種新型脂質(zhì)復(fù)合配方:DLin-MC3-DMA和PEG2000-C-DMG。由于其化學(xué)修飾有限����,Patisiran利用LNP系統(tǒng)在循環(huán)中保持穩(wěn)定,并實(shí)現(xiàn)有效的肝臟攝取����。為了充分包裹LNP,每1毫克劑量的ALN-18328中DLin-MC3-DMA(6.76毫克)和PEG2000-C-DMG(0.76毫克)的用量相當(dāng)����。因此,Patisiran的藥代動(dòng)力學(xué)研究包括了與ALN-18328劑量相關(guān)的DLin-MC3-DMA和PEG2000-C-DMG�。
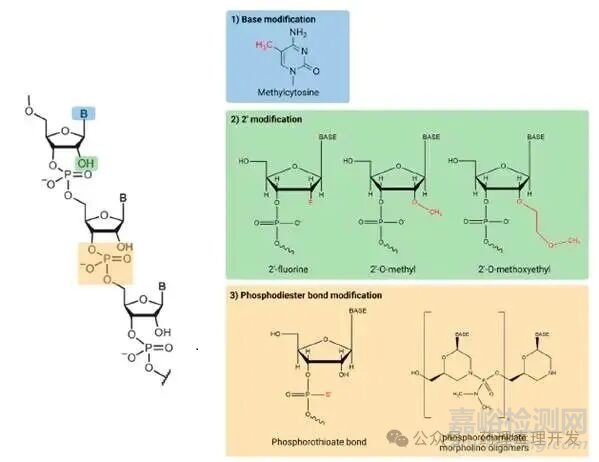
天然寡核苷酸容易被核酸酶迅速降解��,并且對(duì)血漿蛋白親和力較低�,導(dǎo)致其快速被清除。為了克服這些問(wèn)題�,化學(xué)修飾被引入到堿基����、RNA骨架或糖基團(tuán)中����。
首先�,堿基修飾常用于增強(qiáng)沃森-克里克堿基配對(duì)的強(qiáng)度��,這種方法還可以提高寡核苷酸與目標(biāo)RNA形成的雙鏈的熱穩(wěn)定性。然而�,更高的親和力可能會(huì)增加非靶向毒性風(fēng)險(xiǎn)����,從而導(dǎo)致不良反應(yīng)�。此外�,由于修飾后的嘧啶堿基相對(duì)較大�,可能會(huì)阻礙siRNA介導(dǎo)的沉默作用�,因?yàn)樗鼈儠?huì)阻止RNA誘導(dǎo)沉默復(fù)合體(RISC)的正確結(jié)合�。其次��,RNA的核糖修飾是一種廣泛使用的方法����,用于提高對(duì)核酸酶降解的穩(wěn)定性�,從而增加其PK半衰期�。糖基團(tuán)中最常被修飾的位置是其2'-碳原子����。常見(jiàn)的修飾包括2'-O-甲基(2'-O-Me)、2'-氟RNA(2'-F-RNA)和2'-O-甲氧乙基(2'-O-MOE)�。第三��,核酸骨架中的磷酸二酯鍵也是修飾的靶點(diǎn),因?yàn)楹怂崦缚梢运膺@些磷酸二酯鍵��。核酸骨架中最常見(jiàn)的修飾是磷酸硫代鍵(PS)�,其中非橋連氧被硫原子取代。磷酸硫代鍵的優(yōu)勢(shì)不僅在于其對(duì)核酸酶降解的抵抗力,還在于其能夠增加組織攝取����。另一種常見(jiàn)的ASO修飾是磷酸二酰胺嗎啉代寡核苷酸(PMO)�,它包含通過(guò)磷酸二酰胺鍵連接的嗎啉環(huán)修飾的糖�。PMO對(duì)生物體液中的各種酶具有抵抗力����,增強(qiáng)了RNA治療藥物的生物穩(wěn)定性和療效����。
吸收和給藥途徑
RNA藥物通常通過(guò)非口服途徑給藥����,常見(jiàn)的方式是皮下注射或靜脈注射�。與其它藥物相比,RNA藥物皮下給藥后的生物利用度較低至中等�,主要是由于核酸酶的降解作用��。例如,Inclisiran大鼠皮下給藥后的系統(tǒng)生物利用度為35.1%-48.9%����,猴中為24.7%-33.8%。皮下注射后�,這些化合物會(huì)迅速被吸收進(jìn)入系統(tǒng)循環(huán),通常在2-4小時(shí)內(nèi)達(dá)峰��。與靜脈注射相比�,皮下給藥后的血藥濃度在達(dá)到峰值后下降得更慢,這是由于藥物從注射部位持續(xù)吸收所致����。特別是Givosiran����,皮下注射后主要分布至肝臟,盡管其在大鼠和猴子中的生物利用度較低��,但肝臟暴露比值顯著高于靜脈給藥途徑。
Fomivirsen和Pegaptanib用于治療眼部疾病,需要局部給藥以優(yōu)化藥物在靶組織中的作用����,而非全身給藥�。因此��,這兩種藥物均通過(guò)直接玻璃體注射給藥。根據(jù)非臨床研究����,F(xiàn)omivirsen在兔和猴眼中的視網(wǎng)膜濃度分別為3.5 µmol/L和0.88 µmol/L��。此外�,藥代動(dòng)力學(xué)模型預(yù)測(cè)�,人眼中視網(wǎng)膜的藥物峰濃度和谷濃度在每?jī)芍芙o藥165 µg時(shí),范圍為5-0.6 µmol/L�。
由于RNA治療藥物分子量較大且骨架中帶有負(fù)電荷��,無(wú)法穿過(guò)血腦屏障。為了使其在中樞神經(jīng)系統(tǒng)發(fā)揮作用��,用于治療脊髓性肌萎縮癥(SMA)的Nusinersen通過(guò)鞘內(nèi)注射給藥。給藥后,Nusinersen迅速分布在腦脊液中����,與血漿中的較大分布容積(29 L)相比,其腦脊液分布容積較?。?.4 L)�。在猴子中��,Nusinersen在腦脊液中的終末半衰期為3-6個(gè)月,在兒童中為4-6個(gè)月�。藥物的終末半衰期較長(zhǎng)�,支持了每4-6個(gè)月一次的維持給藥頻率。
分布
RNA藥物主要與細(xì)胞內(nèi)的細(xì)胞質(zhì)或細(xì)胞核中的RNA靶點(diǎn)相互作用。然而����,RNA藥物從血液擴(kuò)散到外周組織的能力有限����。即使進(jìn)入組織的RNA還要進(jìn)入細(xì)胞才能發(fā)揮作用。由于寡核苷酸藥物不具有膜通透性��,它們通過(guò)吞噬作用或受體介導(dǎo)的內(nèi)吞作用而不是被動(dòng)擴(kuò)散進(jìn)入細(xì)胞��。以ASOs為例,細(xì)胞攝取過(guò)程大致可以分為兩個(gè)步驟:ASOs吸附到細(xì)胞表面蛋白和內(nèi)化�。許多細(xì)胞表面受體參與ASOs的受體介導(dǎo)內(nèi)吞作用����,包括表皮生長(zhǎng)因子受體、G蛋白偶聯(lián)受體和清道夫受體��。特別是關(guān)于清道夫受體的研究有很多����,發(fā)現(xiàn)它們介導(dǎo)裸ASOs的攝取�,例如stabilin-1和stabilin-2介導(dǎo)PS-ASOs的肝臟攝取,清道夫受體A1介導(dǎo)PMOs的肌肉攝取。內(nèi)化的ASOs需要從內(nèi)體中釋放出來(lái)�,才能與細(xì)胞質(zhì)或細(xì)胞核中的目標(biāo)RNA相互作用。這些細(xì)胞內(nèi)ASO的運(yùn)輸過(guò)程被認(rèn)為受到細(xì)胞質(zhì)和細(xì)胞核區(qū)域中多種蛋白的相互作用調(diào)控����。通過(guò)使用生物素標(biāo)記的PS-ASOs進(jìn)行親和力選擇����,Liang等人鑒定了一組與PS-ASOs相互作用的細(xì)胞內(nèi)蛋白�。最近的一項(xiàng)研究表明�,在早期內(nèi)體途徑中����,Rab5C和早期內(nèi)體抗原1(EEA1)參與了PS-ASOs的運(yùn)輸�,并在stabilin介導(dǎo)的內(nèi)化后促進(jìn)它們的內(nèi)體逃逸;在晚期內(nèi)體途徑中,Rab7A和溶酶體雙磷酸甘油磷脂參與了PS-ASOs的運(yùn)輸�。此外��,為了實(shí)現(xiàn)有效的細(xì)胞內(nèi)化����,需要平衡血漿蛋白結(jié)合��。如果RNA藥物與血漿蛋白結(jié)合過(guò)于緊密�,可能會(huì)阻礙其從全身循環(huán)向組織的分布��。相反����,與血漿蛋白結(jié)合較少的藥物則更容易被清除����,主要由于在血液中的代謝或通過(guò)尿液排泄��。
在RNA藥物中引入PS骨架可以使其對(duì)血漿蛋白的親和力在所有種屬中增加約85%�,從而延長(zhǎng)其半衰期并促進(jìn)其在全身組織中的攝取�。具體來(lái)說(shuō)����,具有PS連接的RNA藥物表現(xiàn)出較高的血漿蛋白結(jié)合率:Givosiran為90%����,Inclisiran為87%,Inotersen為94%��,Lumasiran為85%,Mipomersen為96%����,Nusinersen為94%��。當(dāng)然��,事無(wú)絕對(duì),F(xiàn)omivirsen具有PS連接����,但其在猴或兔玻璃體蛋白中的結(jié)合率僅約為40%��。而基于PMO修飾的ASOs是不帶電的,在人體中的蛋白結(jié)合率較低(6%-17%)����,從而限制了其組織吸收。例如��,PMO類藥物Eteplirsen人血漿蛋白結(jié)合率為6.1%-16.5%。由于血漿蛋白結(jié)合與細(xì)胞攝取和腎小球?yàn)V過(guò)相關(guān),PS-ASOs與PMOs相比具有更持久的組織分布和更慢的尿液排泄特性,這可以通過(guò)它們的蛋白結(jié)合差異來(lái)解釋。
對(duì)于以脂質(zhì)納米顆粒(LNP)形式配制的Patisiran,其血漿蛋白結(jié)合通過(guò)兩種方法進(jìn)行評(píng)估�。在第一種方法中����,Patisiran與血清白蛋白和α1-酸性糖蛋白的結(jié)合率分別為0.46%和2.07%。在另一種方法中�,作為L(zhǎng)NP PEG輔料的PEG2000-C-DMG的血漿蛋白結(jié)合率為約97%�。然而,由于PEG2000-C-DMG在Patisiran中的摩爾比遠(yuǎn)低于其他成分�,因此Patisiran的蛋白結(jié)合率更接近血清白蛋白和α1-酸性糖蛋白的結(jié)果。
大多數(shù)RNA治療藥物主要分布于腎臟和肝臟��,這兩個(gè)器官是主要的排泄和代謝器官��。如上所述�,一些具有GalNAc綴合物的RNA藥物主要靶向肝臟細(xì)胞。此外��,由于肌肉組織的分布低,或者稱為“生物利用度低”�,用于治療DMD等肌肉組織疾病的RNA治療藥物通常設(shè)計(jì)為高活性分子。
代謝
RNA治療藥物的降解通常由外切酶和內(nèi)切酶介導(dǎo)��。一般來(lái)說(shuō)�,未修飾的寡核苷酸會(huì)迅速被3'-外切酶降解。與未修飾的寡核苷酸類似�,一些RNA治療藥物主要通過(guò)3'-外切酶水解裂解進(jìn)行代謝。例如����,Givosiran的主要代謝產(chǎn)物AS(N-1) 3' Givosiran是由其反義鏈的3'端降解形成的,并且這種活性代謝產(chǎn)物在血漿中濃度還挺高��。因此����,Givosiran的臨床PK研究測(cè)量了血漿中AS(N-1) 3' Givosiran的暴露水平,其血藥濃度大約是Givosiran的一半。其他siRNA藥物�,如Lumasiran和Inclisiran,也主要代謝為其各自的AS(N-1) 3'代謝產(chǎn)物�,但它們各自形成的代謝產(chǎn)物量遠(yuǎn)低于Givosiran�。Nusinersen同樣通過(guò)3'-外切酶代謝����,其N-1代謝產(chǎn)物在腦脊液和血漿中均被檢測(cè)到。除了3'-外切酶外,RNA藥物的代謝中還觀察到了5'-外切酶和內(nèi)切酶的作用。例如����,Mipomersen是通過(guò)內(nèi)切酶而非外切酶降解的�,經(jīng)Mipomersen治療的患者尿液樣本中檢測(cè)到了與內(nèi)切酶介導(dǎo)裂解相關(guān)的幾種代謝產(chǎn)物。
肝臟是大多數(shù)藥物代謝的場(chǎng)所�,因此RNA藥物也在肝臟的S9組分、肝微粒體或重組細(xì)胞色素P450(CYP)系統(tǒng)中進(jìn)行了測(cè)試�,以了解每種藥物的代謝情況。大多數(shù)GalNAc siRNA藥物不是CYP酶的底物��,因此不太可能發(fā)生涉及CYP酶的臨床相互作用�。此外,包括Mipomersen����、Eteplirsen、Nusinersen和Inotersen在內(nèi)的幾種ASO藥物也不經(jīng)CYP酶介導(dǎo)的代謝�。
RNA藥物的代謝產(chǎn)物主要是通過(guò)特定酶(如外切酶或內(nèi)切酶)形成的親本寡核苷酸片段,非臨床研究可以作為人體放射性標(biāo)記研究的替代方法�。可以通過(guò)體外研究或動(dòng)物物料平衡研究獲得代謝相關(guān)信息�。例如��,Eteplirsen開(kāi)展了小鼠放射性標(biāo)記研究�,而沒(méi)有進(jìn)行人體放射性標(biāo)記研究�。Lumasiran的情況也類似,主要采用非臨床數(shù)據(jù)對(duì)代謝情況進(jìn)行了研究��。
排泄
盡管腎臟是大多數(shù)RNA治療藥物的主要排泄途徑�,但每種RNA藥物都有與其排泄相關(guān)的特定PK參數(shù)。例如��,F(xiàn)omivirsen從玻璃體中的清除速率呈一級(jí)動(dòng)力學(xué)��,其半衰期約為55小時(shí)�。玻璃體中Fomivirsen濃度的時(shí)間依賴性降低可能是由于其被視網(wǎng)膜和其他眼部組織攝取,或被核酸酶代謝����。此外,Mipomersen在單次給藥后24小時(shí)內(nèi)尿液排泄量極少����,僅為1.38%至3.30%�。由于Mipomersen在人體中的半衰期為31天,其清除過(guò)程延遲��,表明其在組織中代謝緩慢,隨后通過(guò)尿液排泄�。Inotersen與血漿蛋白結(jié)合緊密,因此其通過(guò)腎小球?yàn)V過(guò)的量極少����。在給藥后24小時(shí)內(nèi),完整Inotersen的腎臟排泄量不到給藥劑量的1%����。Inotersen的消除半衰期為32.3天,表明其在早期分布階段后的代謝或排泄過(guò)程較長(zhǎng)����。Nusinersen也通過(guò)腎臟排泄,其代謝產(chǎn)物是核酸酶降解產(chǎn)物��。Nusinersen通過(guò)鞘內(nèi)注射給藥��,必須進(jìn)入血液才能通過(guò)腎臟排泄����。然而,由于腦脊液與血液之間的屏障�,Nusinersen的消除半衰期可能會(huì)延長(zhǎng)。
所有獲得FDA批準(zhǔn)的PMO類RNA治療藥物(包括Casimersen��、Eteplirsen、Golodirsen和Viltolarsen)均以原形藥物的形式迅速通過(guò)尿液排泄�。這些藥物在24小時(shí)內(nèi)尿液中的回收率均超過(guò)60%。它們的消除半衰期和血漿清除率分別為:Casimersen為3.5小時(shí)和180 mL/h/kg��,Eteplirsen為1.6-3.6小時(shí)和200-300 mL/h/kg�,Golodirsen為3.5小時(shí)和338-405 mL/h/kg,Viltolarsen為2.5小時(shí)和217 mL/h/kg����。
GalNAc綴合的siRNA藥物(如Givosiran、Inclisiran�、Lumasiran和Vutrisiran)已獲得批準(zhǔn)?;诜桥R床數(shù)據(jù),siRNA藥物的初始血液清除主要是通過(guò)在肝臟中的高分布實(shí)現(xiàn)的�。Givosiran通過(guò)轉(zhuǎn)化為AS(N-1) 3' Givosiran(36%)、在肝臟中的攝?。?2%)和尿液排泄(12%)從血漿中清除。相比之下�,Inclisiran的肝臟攝取介導(dǎo)的清除更高,約82.5%����。此外,Lumasiran的大部分也被肝臟攝取�,僅有17.4%至25.8%的給藥劑量以原形藥物的形式通過(guò)尿液排泄。Vutrisiran的尿液排泄量不到給藥總量的25%��。然而��,GalNAc綴合siRNA藥物在肝臟中的消除半衰期較長(zhǎng)��,例如Inclisiran(猴)為82.5天��,Lumasiran(基于PBPK模型預(yù)測(cè))為66.9天�。這些緩慢的消除速率表明,這些藥物的糞便排泄量可能較少����,主要排泄途徑可能是腎臟。
為了研究腎功能是否會(huì)影響GalNAc綴合siRNA的PK特征��、腎臟排泄或藥效學(xué)(PD)結(jié)果����,進(jìn)行了5/6腎切除大鼠研究。在代表中度至重度腎功能不全的5/6腎切除大鼠模型中��,肝臟的PK特征或觀察到的PD結(jié)果并未因尿液排泄減少而受到顯著影響����。因此�,腎功能不全不太可能影響GalNAc綴合siRNA的肝臟PK特征及其隨后的PD結(jié)果�。類似的結(jié)果也在腎功能不全患者的Inclisiran臨床研究中發(fā)現(xiàn)。對(duì)于Givosiran和Lumasiran��,亞組分析結(jié)果顯示��,輕度至中度腎功能不全似乎不會(huì)影響療效�。
藥物-藥物相互作用
大多數(shù)RNA類治療藥物不是細(xì)胞色素P450(CYP)系統(tǒng)的抑制劑或誘導(dǎo)劑,因此通常認(rèn)為它們不會(huì)與主要通過(guò)氧化代謝途徑清除的小分子藥物發(fā)生經(jīng)典的藥–藥相互作用����。然而,已有臨床研究報(bào)道了givosiran可能引發(fā)間接的藥–藥相互作用��。該研究指出����,由于givosiran的藥效學(xué)作用,可能影響肝臟中的血紅素含量——而血紅素是CYP酶的輔基和功能所必需的輔因子����。研究結(jié)果顯示:givosiran對(duì)CYP2C9活性幾乎沒(méi)有影響;對(duì)CYP3A4和CYP2C19活性有輕微抑制��;對(duì)CYP2D6和CYP1A2活性則呈現(xiàn)中度抑制。值得注意的是��,體外使用人肝微粒體的研究表明��,givosiran并未直接抑制任何主要CYP亞型�,這可能是因?yàn)槠渌幮W(xué)特性難以在體外模型中充分展現(xiàn)��。
除了代謝酶��,轉(zhuǎn)運(yùn)體也能通過(guò)調(diào)節(jié)藥物的吸收�、分布和消除,對(duì)藥代動(dòng)力學(xué)和藥效學(xué)產(chǎn)生臨床相關(guān)影響�。與主要在肝臟和小腸表達(dá)的藥物代謝酶不同,轉(zhuǎn)運(yùn)體廣泛分布于全身��,調(diào)控物質(zhì)在胃腸道����、肝臟、腎臟和大腦等組織間的轉(zhuǎn)運(yùn)�。因此,在部分RNA治療藥物的研發(fā)過(guò)程中����,已對(duì)其經(jīng)由轉(zhuǎn)運(yùn)體介導(dǎo)的藥–藥相互作用潛力進(jìn)行了評(píng)估。
大多數(shù)GalNAc偶聯(lián)的siRNA藥物被證實(shí)不是臨床相關(guān)轉(zhuǎn)運(yùn)體的底物,且不抑制關(guān)鍵的攝取轉(zhuǎn)運(yùn)體(如OATP1B1����、OATP1B3、OAT3�、OCT1、OCT2)或外排轉(zhuǎn)運(yùn)體(如BCRP����、BSEP、MATE1����、MATE2-K、P-gp)��,但givosiran在高濃度下可抑制P-gp����。然而,givosiran采用皮下給藥��,其體內(nèi)濃度與體外IC50比值(I/ IC50)遠(yuǎn)低于Ellens和Lee提出的臨床顯著性閾值(0.03)�,因此該P(yáng)-gp抑制作用無(wú)臨床意義。
此外�,多種ASO藥物(如casimersen�、eteplirsen����、golodirsen、inotersen��、mipomersen�、nusinersen和viltolarsen)也已在體外評(píng)估了其與轉(zhuǎn)運(yùn)體相關(guān)的藥代動(dòng)力學(xué)相互作用。結(jié)果一致表明:這些ASO藥物既非轉(zhuǎn)運(yùn)體底物����,也不抑制多種人體轉(zhuǎn)運(yùn)體��,因此不太可能因競(jìng)爭(zhēng)或抑制而與其他藥物發(fā)生相互作用����。
關(guān)于血漿蛋白結(jié)合。盡管部分RNA治療藥物具有較高的血漿蛋白結(jié)合率��,理論上存在因蛋白結(jié)合位點(diǎn)競(jìng)爭(zhēng)而導(dǎo)致藥代動(dòng)力學(xué)相互作用的可能[83]�,但實(shí)際上這種風(fēng)險(xiǎn)極低,原因如下:1)給藥頻率低(通常每月一次或更長(zhǎng))����,且藥物在血液循環(huán)中僅短暫存在(數(shù)小時(shí)),隨后迅速分布至組織,因此血漿蛋白結(jié)合的影響有限�;2)臨床峰濃度遠(yuǎn)低于血漿蛋白(如白蛋白,濃度約600μM)水平����;3)寡核苷酸與血漿蛋白的結(jié)合較弱,且其親水性結(jié)構(gòu)所結(jié)合的位點(diǎn)通常不同于疏水性小分子藥物的結(jié)合位點(diǎn)����。綜上,血漿蛋白結(jié)合對(duì)RNA治療藥物的藥–藥相互作用影響甚微�,基本可忽略。
PK/PD分析將藥物劑量–濃度關(guān)系與藥物濃度–效應(yīng)關(guān)系聯(lián)系起來(lái)��,從而預(yù)測(cè)隨時(shí)間變化的給藥效果�。在PD分析中,常使用生物標(biāo)志物來(lái)評(píng)估給藥方案與靶點(diǎn)效應(yīng)之間的關(guān)聯(lián)�。
由于RNA治療藥物旨在特異性作用于特定靶基因,其主要的PD生物標(biāo)志物通常較為明確����。例如:Inotersen靶向轉(zhuǎn)甲狀腺素蛋白(TTR)mRNA,其PD生物標(biāo)志物為血漿TTR蛋白水平的變化�;Inclisiran是一種針對(duì)高脂血癥相關(guān)蛋白PCSK9設(shè)計(jì)的化學(xué)合成siRNA,其PD生物標(biāo)志物為PCSK9蛋白濃度����。
不過(guò)��,盡管PD生物標(biāo)志物明確����,但RNA治療藥物的PK/PD關(guān)系比傳統(tǒng)小分子藥物更為復(fù)雜��。這種復(fù)雜性在生物藥物中也普遍存在��,已有研究提出靶點(diǎn)介導(dǎo)的藥物處置模型(Target-Mediated Drug Disposition, TMDD)來(lái)解釋此類現(xiàn)象����。近年來(lái)�,該模型也被應(yīng)用于RNA治療藥物的PK/PD建模。
RNA治療藥物的一個(gè)顯著特點(diǎn)是血漿中的PK參數(shù)與PD效應(yīng)之間缺乏直接相關(guān)性��。以vutrisiran為例��,在健康志愿者中����,其血漿PK Tmax為0.2–12小時(shí),而作為PD指標(biāo)的TTR最低水平(即PD Tmax)卻出現(xiàn)在給藥后50–90天����。另外一個(gè)特點(diǎn)是血漿半衰期短��,但藥效持續(xù)時(shí)間長(zhǎng)�。例如����,用于DMD治療的ASO藥物血漿半衰期通常小于4小時(shí),但每周一次靜脈給藥即可維持臨床療效��。這說(shuō)明即使藥物迅速?gòu)难獫{中清除�,其藥理作用仍可持續(xù)數(shù)周甚至數(shù)月。
為解釋血漿PK與PD效應(yīng)之間的脫節(jié)����,需考察其他藥代動(dòng)力學(xué)特征。研究發(fā)現(xiàn)當(dāng)RNA治療藥物具有明確靶器官時(shí)��,其PD效應(yīng)與靶器官內(nèi)的藥物濃度相關(guān)性更強(qiáng)�,而非血漿濃度;對(duì)于siRNA類藥物����,在非臨床研究中�,其PD效應(yīng)更直接地與RISC復(fù)合物(RNA誘導(dǎo)沉默復(fù)合體)相關(guān)����,因?yàn)镽ISC是siRNA發(fā)揮功能的核心結(jié)構(gòu)。
因此�,目前RNA治療藥物的濃度–效應(yīng)關(guān)系尚未完全闡明��。
群體藥代動(dòng)力學(xué)分析
過(guò)去十年中��,群體藥代動(dòng)力學(xué)(PopPK)或PK/PD建模與模擬已被應(yīng)用于部分獲批RNA治療藥物的臨床評(píng)價(jià)����。
然而,在杜氏肌營(yíng)養(yǎng)不良癥(DMD)治療藥物的開(kāi)發(fā)中�,由于DMD屬于超罕見(jiàn)病,且多數(shù)患者年齡較小����、難以采集足夠樣本進(jìn)行有效分析�,因此相關(guān)藥物獲批時(shí)往往僅基于有限的PopPK數(shù)據(jù),甚至未建立PK/PD模型�。
PopPK方法的優(yōu)勢(shì)在于:能整合密集采樣或稀疏采樣的藥代動(dòng)力學(xué)數(shù)據(jù);可構(gòu)建模型以解釋個(gè)體間變異(如年齡����、體重�、肝腎功能����、基因型等);有助于優(yōu)化給藥方案的選擇��。
因此�,PopPK方法特別適用于由遺傳缺陷引起的罕見(jiàn)病的RNA治療藥物研發(fā)—這類疾病不僅患者招募困難����,且受試者年齡跨度大(包括兒童)��。
目前已有兩項(xiàng)公開(kāi)發(fā)表的群體PK/PD研究����,分別針對(duì)治療相同疾?�。ㄟz傳性轉(zhuǎn)甲狀腺素蛋白淀粉樣變性��,hATTR)的兩種RNA藥物:inotersen(ASO)和patisiran(siRNA)�。
1.Inotersen(反義寡核苷酸)
基于I����、II����、III期臨床試驗(yàn)數(shù)據(jù)建立了群體PK/PD模型。其PK/PD關(guān)系相對(duì)明確��,因inotersen半衰期較長(zhǎng)�,可使用兩室模型將血漿PK與TTR蛋白水平有效關(guān)聯(lián)。
分析顯示:體重或體型是影響清除率和分布容積的顯著協(xié)變量�,但對(duì)藥物暴露量無(wú)臨床相關(guān)影響;輕中度腎功能不全患者的腎功能不是清除率的顯著協(xié)變量����。
模型模擬了四種給藥方案:300mg每周一次(±負(fù)荷劑量);300mg每?jī)芍芤淮危?50mg每周一次��。結(jié)果提示較低劑量同樣有效����,且長(zhǎng)期治療中負(fù)荷劑量在3個(gè)月后作用甚微�,可能無(wú)需使用�。
2.Patisiran(siRNA�,首個(gè)建立群體PK/PD模型的siRNA藥物)
該模型通過(guò)將血漿藥物濃度與肝細(xì)胞內(nèi)RISC復(fù)合物水平關(guān)聯(lián)��,成功建立了PK/PD關(guān)系�。臨床觀察到藥效峰值滯后于血漿濃度峰值7–14天。
模型引入獨(dú)立效應(yīng)室(effect compartment)描述這種滯后現(xiàn)象(hysteresis)��,并能準(zhǔn)確擬合不同給藥方案下TTR的時(shí)間變化曲線����。
模擬結(jié)果表明,患者基因型對(duì)藥效無(wú)顯著影響��。支持當(dāng)前獲批方案�,體重<100kg,0.3mg/kg每三周一次(Q3W)��。在體重36.2–110kg范圍內(nèi)����,即使PK暴露量增加一倍,藥效未見(jiàn)明顯差異��。
對(duì)于體重≥100kg的患者,模擬顯示:固定劑量30mg Q3W與按體重計(jì)算的0.3mg/kg Q3W所產(chǎn)生的藥效相似����。因此,30mg可作為最大劑量上限用于100kg及以上患者����。
免疫原性
以siRNA為例����,siRNA可以誘導(dǎo)先天免疫系統(tǒng)�,且siRNA和遞送系統(tǒng)也可能有一定的免疫原性風(fēng)險(xiǎn)。所以��,siRNA需要考察ADA產(chǎn)生情況�。不過(guò),目前獲批的6款siRNA的ADA風(fēng)險(xiǎn)都很低����,patisiran、givosiran�、lumasiran����、inclisiran��、vutrisiran��、nedosiran的臨床ADA發(fā)生率分別為3.6%�、0.9%�、6%、1.7%��、2.5%�、0%。而且�,產(chǎn)生的ADA對(duì)藥物的PK、PD��、有效性或安全性均未產(chǎn)生影響��。