摘要:抗體藥物憑借其高特異性與靶向性����、低毒性和可工程化特性等優(yōu)勢(shì)已成為生物醫(yī)藥領(lǐng)域的核心治療手段, 在腫瘤���、自身免疫性疾病�、感染性疾病預(yù)防/治療���、心血管代謝病�、眼科疾病等領(lǐng)域獲得巨大成功. 盡管雜交瘤技術(shù)、噬菌體展示技術(shù)�、酵母展示技術(shù)等傳統(tǒng)抗體發(fā)現(xiàn)技術(shù)已成功推動(dòng)了大量抗體藥物的研發(fā)與上市, 但這些方法存在研發(fā)周期長(zhǎng)、篩選效率低����、對(duì)復(fù)雜抗原響應(yīng)能力弱等固有局限. 近年來(lái), 人工智能技術(shù)的飛速發(fā)展, 尤其是深度學(xué)習(xí)在蛋白質(zhì)結(jié)構(gòu)預(yù)測(cè)和序列建模方面的突破, 為抗體藥物的設(shè)計(jì)注入了前所未有的活力. 人工智能不僅顯著提升了抗體發(fā)現(xiàn)的效率, 為突發(fā)公共衛(wèi)生事件下抗體藥物的研發(fā)提供了關(guān)鍵技術(shù)支持, 更為個(gè)體化精準(zhǔn)治療提供了可擴(kuò)展、可調(diào)控的技術(shù)支持. 本文系統(tǒng)梳理了人工智能在抗體藥物設(shè)計(jì)全流程中的應(yīng)用, 涵蓋從序列生成���、結(jié)構(gòu)建模到親和力預(yù)測(cè)與優(yōu)化成熟等環(huán)節(jié), 并探討了當(dāng)前挑戰(zhàn)與未來(lái)發(fā)展方向���。
隨著新時(shí)代藥物研發(fā)對(duì)抗體性能(如高特異性、低免疫原性���、復(fù)雜靶點(diǎn)結(jié)合能力)的要求提升, 傳統(tǒng)抗體發(fā)現(xiàn)技術(shù)(如雜交瘤技術(shù)����、噬菌體展示技術(shù)����、酵母展示技術(shù)等)的固有局限性日益凸顯, 例如, 傳統(tǒng)抗體發(fā)現(xiàn)技術(shù)依賴實(shí)驗(yàn)篩選, 研發(fā)周期長(zhǎng)且成本高昂. 此外, 傳統(tǒng)抗體發(fā)現(xiàn)技術(shù)的原理(如依賴動(dòng)物免疫應(yīng)答、簡(jiǎn)單體外展示系統(tǒng))與復(fù)雜抗原的特性(構(gòu)象依賴、免疫耐受�、動(dòng)態(tài)變化)存在核心沖突, 因此導(dǎo)致傳統(tǒng)技術(shù)對(duì)復(fù)雜抗原的抗體發(fā)現(xiàn)效率極低, 而這類抗原可能剛好是腫瘤、自身免疫疾病���、感染性疾病的關(guān)鍵靶點(diǎn).
隨著高通量測(cè)序技術(shù)在抗體篩選中的廣泛應(yīng)用,顯著拓展了抗體序列多樣性的認(rèn)知. 抗體鏈間殘基的相互作用機(jī)制也隨之得到更深入的解析. 與此同時(shí),人工智能(artificial intelligence, AI)技術(shù)尤其是深度學(xué)習(xí)模型在蛋白質(zhì)結(jié)構(gòu)預(yù)測(cè)和序列建模方面的突破, 為抗體藥物設(shè)計(jì)賦予了新的動(dòng)能 [1,2]. AI不僅顯著提升了抗體發(fā)現(xiàn)的效率, 更關(guān)鍵的是, 其為個(gè)體化精準(zhǔn)治療構(gòu)建了兼具可擴(kuò)展性與可調(diào)控性的技術(shù)支撐體系 [3~5]. 目前, 通過(guò)多模態(tài)模型融合與動(dòng)態(tài)數(shù)據(jù)反饋等對(duì)抗體研發(fā)邏輯進(jìn)行深度重構(gòu), 已逐步構(gòu)建起一個(gè)多層次�、端到端的抗體設(shè)計(jì)框架, 基于AI的抗體優(yōu)化路徑已經(jīng)初步實(shí)現(xiàn)端到端, 從序列生成���、結(jié)構(gòu)建模, 到親和力預(yù)測(cè)與成熟, 構(gòu)建起完整的智能抗體設(shè)計(jì)流程 [6,7]. 盡管AI技術(shù)在抗體藥物設(shè)計(jì)領(lǐng)域取得了革命性突破, 但在數(shù)據(jù)的質(zhì)量和數(shù)量����、模型的可解釋性等方面仍面臨亟須解決的核心瓶頸. 本文系統(tǒng)梳理了人工智能在抗體藥物設(shè)計(jì)全流程中的應(yīng)用, 涵蓋從序列生成����、結(jié)構(gòu)建模到親和力預(yù)測(cè)與優(yōu)化成熟等環(huán)節(jié), 并探討了當(dāng)前挑戰(zhàn)與未來(lái)發(fā)展方向.
1 概述
在抗體藥物設(shè)計(jì)領(lǐng)域, AI技術(shù)的融入正深刻改變傳統(tǒng)依賴實(shí)驗(yàn)篩選的范式, 轉(zhuǎn)而通過(guò)計(jì)算模擬實(shí)現(xiàn)高效、精準(zhǔn)的分子優(yōu)化. 抗體藥物設(shè)計(jì)的本質(zhì)在于識(shí)別并強(qiáng)化抗原-抗體結(jié)合界面, 其中互補(bǔ)位(paratope)與表位(epitope)的精準(zhǔn)建模至關(guān)重要. 近年來(lái), 卷積神經(jīng)網(wǎng)絡(luò)(convolutional neural networks, CNN)����、循環(huán)神經(jīng)網(wǎng)絡(luò)(recurrent neural network, RNN)Transformer和擴(kuò)散模型(diffusion models)等一系列模型推動(dòng)了端到端的抗體智能設(shè)計(jì)框架形成, 這些模型既能捕捉序列的局部/全局依賴, 也能聯(lián)動(dòng)三維結(jié)構(gòu)與功能. CNN作為最早應(yīng)用于抗體設(shè)計(jì)的深度學(xué)習(xí)模型之一, 主要用于提取序列或結(jié)構(gòu)中的局部特征 [8]. 在抗體序列分析中,CNN可識(shí)別CDR區(qū)域的氨基酸模式, 幫助預(yù)測(cè)結(jié)合位點(diǎn)或突變影響. 例如, CNN模型已被用于表位預(yù)測(cè), 通過(guò)卷積層掃描抗原序列的連續(xù)片段, 識(shí)別潛在免疫原位點(diǎn) [9]. CNN的優(yōu)勢(shì)在于計(jì)算效率高, 能處理高維數(shù)據(jù), 并易于并行化; 然而, CNN忽略長(zhǎng)距離依賴, 無(wú)法捕捉抗體重鏈與輕鏈間的全局交互, 導(dǎo)致在復(fù)雜構(gòu)象預(yù)測(cè)中的準(zhǔn)確率較低, 尤其對(duì)CDR-H3環(huán)的變異性建模不足. RNN及長(zhǎng)短期記憶網(wǎng)絡(luò)(long short-term memory,LSTM), 則擅長(zhǎng)處理序列數(shù)據(jù)的時(shí)序依賴, 適用于抗體序列生成和親和力成熟模擬 [10,11]. Transformer [12]的出現(xiàn)則標(biāo)志著抗體設(shè)計(jì)進(jìn)入高效全局建模時(shí)代, 其自注意力機(jī)制允許并行捕捉序列中的長(zhǎng)距離關(guān)系. 如AlphaFold2, 其Transformer-based Evoformer模塊整合多序列比對(duì)(multiple sequence alignment, MSA), 實(shí)現(xiàn)高精度抗體結(jié)構(gòu)預(yù)測(cè) [13]. 在抗體設(shè)計(jì)中, Transformer用于序列嵌入和結(jié)構(gòu)生成, 如AntiBERTy [14]通過(guò)掩碼訓(xùn)練捕捉抗體進(jìn)化信號(hào), 支持親和力優(yōu)化. Transformer的優(yōu)勢(shì)包括并行計(jì)算、處理長(zhǎng)序列的卓越性能,以及多模態(tài)融合能力; 在計(jì)算層面, 自注意力機(jī)制在序列上呈二次復(fù)雜度, 限制了長(zhǎng)鏈或多體建模與大批高通量篩選的成本效率 [15,16]; 在可解釋性與泛化層面,注意力權(quán)重不完全等同于因果解釋這一點(diǎn)在通用自然語(yǔ)言模型中已被檢驗(yàn) [17], 這意味著抗體場(chǎng)景下需要配套的歸因與物理校驗(yàn). 局限在于高計(jì)算資源需求和對(duì)訓(xùn)練數(shù)據(jù)量的依賴, 在有限樣本數(shù)據(jù)集上容易產(chǎn)生過(guò)擬合的問(wèn)題, 影響模型的準(zhǔn)確性和泛化能力 [18]. 擴(kuò)散模型作為生成式AI的最新模型, 通過(guò)逐步添加/去除噪聲模擬分子動(dòng)態(tài), 用于抗體序列或結(jié)構(gòu)從頭設(shè)計(jì) [19].RFdiffusion-Antibody利用擴(kuò)散過(guò)程生成目標(biāo)特異性抗體框架, 支持多表位優(yōu)化. 優(yōu)勢(shì)在于產(chǎn)生多樣化�、高質(zhì)量候選, 提升設(shè)計(jì)創(chuàng)新性; 缺點(diǎn)是訓(xùn)練復(fù)雜、推理時(shí)間長(zhǎng), 且對(duì)噪聲調(diào)度敏感. 這些模型的優(yōu)缺點(diǎn)互補(bǔ), 推動(dòng)抗體設(shè)計(jì)向智能化演進(jìn): CNN/RNN處理局部序列,Transformer提供全局視角, 擴(kuò)散模型注入創(chuàng)造性. 未來(lái), 通過(guò)集成框架如多模態(tài)Transformer-Diffusion融合 [19], 將進(jìn)一步提升抗體親和力和穩(wěn)定性, 加速?gòu)挠?jì)算預(yù)測(cè)到臨床轉(zhuǎn)化的進(jìn)程.
為直觀體現(xiàn)傳統(tǒng)技術(shù)與AI路線的差異, 本文進(jìn)行了示例性對(duì)比(見表1, 數(shù)據(jù)區(qū)間取自公開報(bào)道與行業(yè)實(shí)踐的常見量級(jí), 僅作范式說(shuō)明, 不構(gòu)成統(tǒng)一標(biāo)準(zhǔn); 具體數(shù)值應(yīng)隨項(xiàng)目與實(shí)驗(yàn)體系校準(zhǔn)).
2 人工智能賦能的抗體設(shè)計(jì)流程
2.1 抗體序列分析與生成
利用AI技術(shù)對(duì)大量抗體序列數(shù)據(jù)進(jìn)行分析, 可以揭示抗體序列的特征和規(guī)律, 如氨基酸組成����、序列保守性、序列多樣性等. 這些信息對(duì)于理解抗體的功能和結(jié)構(gòu)具有重要意義. 近年來(lái)涌現(xiàn)出多種基于蛋白語(yǔ)言模型(protein language models, PLMs)的AI技術(shù), 這些模型在捕捉抗體序列中的上下文依賴性�、突變影響及功能注釋方面展現(xiàn)出卓越性能. 在序列生成與區(qū)域重構(gòu)方面, nanoBERT [20]是基于BERT(bidirectional encoder representations from transformers)架構(gòu)、專為納米抗體序列分析與優(yōu)化設(shè)計(jì)的深度學(xué)習(xí)模型. 該模型以INDI數(shù)據(jù)庫(kù) [21]中千萬(wàn)級(jí)別的納米抗體序列作為訓(xùn)練數(shù)據(jù), 能夠在無(wú)基因標(biāo)簽的前提下, 預(yù)測(cè)特定位點(diǎn)的氨基酸替換, 并生成新型的互補(bǔ)決定區(qū)(complementarity- determining region, CDR)序列. 研究結(jié)果表明, 在V區(qū)序列重建任務(wù)中, nanoBERT相較于以人源抗體訓(xùn)練的模型高約12個(gè)百分點(diǎn). IgLM [22]是另一種具有代表性的模型, 基于GPT-2架構(gòu)構(gòu)建, 支持對(duì)抗體序列中指定區(qū)域的插空式生成(inpainting), 并能根據(jù)不同物種和輕/重鏈類型靈活調(diào)整設(shè)計(jì)策略, 增強(qiáng)模型適應(yīng)性. 與此同時(shí), 為了捕捉抗體序列的進(jìn)化邏輯與填補(bǔ)高通量測(cè)序中的缺失殘基, 研究人員又提出了AntiBERTy [14]和AbLang [23]兩個(gè)模型. AntiBERTy是一種基于BERT架構(gòu)的抗體語(yǔ)言模型, 它在超過(guò)5.58億條人源抗體重鏈和輕鏈序列上進(jìn)行掩碼語(yǔ)言預(yù)訓(xùn)練, 該模型的嵌入不僅能重現(xiàn)抗體定向進(jìn)化路徑, 還可精準(zhǔn)識(shí)別關(guān)鍵結(jié)合殘基, 為下游表位預(yù)測(cè)和親和力評(píng)估提供了高效且富有生物學(xué)意義的特征 [24]. AbLang則利用ObservedAntibody Space(OAS)數(shù)據(jù)庫(kù)中的大規(guī)模B細(xì)胞受體序列進(jìn)行訓(xùn)練 [23], 無(wú)須依賴任何種系序列信息, 即可以遠(yuǎn)快于通用模型的速度準(zhǔn)確重構(gòu)完整抗體序列, 從而在序列重構(gòu)���、突變體設(shè)計(jì)及后續(xù)結(jié)構(gòu)預(yù)測(cè)等應(yīng)用場(chǎng)景中表現(xiàn)出顯著優(yōu)勢(shì). 此外, 作為通用蛋白語(yǔ)言模型的代表, ESM-2 [25]具備出色的序列嵌入能力, 可用于支持蛋白結(jié)構(gòu)預(yù)測(cè)���、穩(wěn)定性分析與功能注釋等任務(wù). 在抗體結(jié)合位點(diǎn)預(yù)測(cè)任務(wù)中, ParaAntiProt [26]模型將ESM-2作為特征編碼器, 顯著提升了抗體結(jié)合位點(diǎn)的預(yù)測(cè)性能.具體見表2.
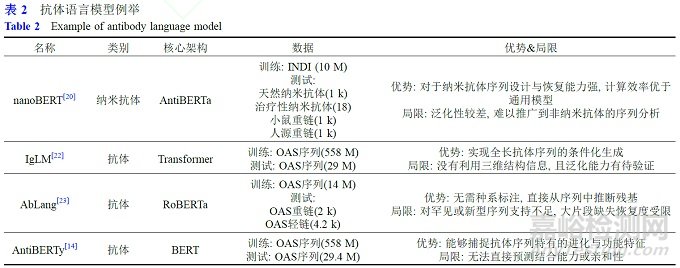
這些AI模型憑借對(duì)抗體序列上下文語(yǔ)義的建模能力和對(duì)結(jié)構(gòu)功能關(guān)系的高效捕捉, 不僅顯著提升了抗體序列生成、結(jié)合位點(diǎn)識(shí)別及親和力優(yōu)化的效率與精度, 也推動(dòng)了從納米抗體到高親和力突變體的系統(tǒng)性設(shè)計(jì)流程. 它們的應(yīng)用標(biāo)志著抗體工程正邁入一個(gè)由大規(guī)模數(shù)據(jù)驅(qū)動(dòng)�、深度模型引領(lǐng)的智能設(shè)計(jì)新時(shí)代.
2.2 抗體結(jié)構(gòu)預(yù)測(cè)
抗體的結(jié)構(gòu)與其功能密切相關(guān), 準(zhǔn)確的抗體結(jié)構(gòu)預(yù)測(cè)對(duì)于理解抗體的抗原識(shí)別機(jī)制、設(shè)計(jì)高親和力抗體以及開發(fā)基于結(jié)構(gòu)的藥物具有重要意義. 傳統(tǒng)的抗體結(jié)構(gòu)預(yù)測(cè)方法主要依賴于實(shí)驗(yàn)技術(shù), 如X射線晶體學(xué)和核磁共振等, 但這些方法存在耗時(shí)長(zhǎng)����、成本高、對(duì)樣品要求高等問(wèn)題. 在抗體三維結(jié)構(gòu)預(yù)測(cè)領(lǐng)域, AI模型正持續(xù)突破傳統(tǒng)技術(shù)瓶頸, 特別是在結(jié)構(gòu)高度可變的CDR區(qū)域, 尤其是CDR-H3環(huán)的建模方面展現(xiàn)出顯著優(yōu)勢(shì). AlphaFold2 [13]和AlphaFold3 [27]是當(dāng)前主流的結(jié)構(gòu)預(yù)測(cè)模型, 均基于深度神經(jīng)網(wǎng)絡(luò)(deep neuralnetwork, DNN)架構(gòu), 能夠僅憑蛋白質(zhì)序列信息預(yù)測(cè)其三維結(jié)構(gòu). 其中, AlphaFold2在第十四屆蛋白質(zhì)結(jié)構(gòu)預(yù)測(cè)評(píng)估競(jìng)賽(CASP14)中表現(xiàn)卓越, 推動(dòng)了結(jié)構(gòu)生物學(xué)的重大變革, 而AlphaFold3在此基礎(chǔ)上進(jìn)一步引入擴(kuò)散模塊, 以提升多鏈復(fù)合物的結(jié)構(gòu)預(yù)測(cè)精度. 然而, AlphaFold系列模型雖在通用蛋白質(zhì)預(yù)測(cè)任務(wù)中取得巨大成功, 但在應(yīng)對(duì)抗體特有的結(jié)構(gòu)多樣性和免疫功能關(guān)聯(lián)性方面仍存在局限, 尤其是難以精準(zhǔn)刻畫CDR區(qū)域的細(xì)節(jié)結(jié)構(gòu). 為彌補(bǔ)通用結(jié)構(gòu)預(yù)測(cè)模型在抗體特異性建模方面的不足, 研究者開始開發(fā)面向抗體序列與結(jié)構(gòu)特點(diǎn)的專用結(jié)構(gòu)預(yù)測(cè)工具. DeepAb [28]是較早應(yīng)用于抗體結(jié)構(gòu)建模的深度學(xué)習(xí)方法之一, 采用兩階段策略對(duì)Fv區(qū)結(jié)構(gòu)進(jìn)行預(yù)測(cè). 該模型首先從重鏈和輕鏈序列中預(yù)測(cè)殘基之間的幾何約束信息(包括距離和角度), 隨后將這些預(yù)測(cè)結(jié)果輸入Rosetta進(jìn)行三維結(jié)構(gòu)生成. 與傳統(tǒng)方法相比, DeepAb引入了具備可解釋性的注意力機(jī)制, 能夠識(shí)別并突出結(jié)構(gòu)中如芳香堆疊或氫鍵網(wǎng)絡(luò)等關(guān)鍵殘基對(duì)的作用, 從而在多個(gè)抗體測(cè)試集中表現(xiàn)出穩(wěn)定的建模能力. 然而, 該方法依賴外部建模工具���、預(yù)測(cè)速度較慢, 且不具備端到端結(jié)構(gòu)生成能力, 因此在高通量應(yīng)用場(chǎng)景中存在一定局限. 為提高建模效率并簡(jiǎn)化流程, 研究者提出了ABlooper模型 [29].該模型聚焦于抗體CDR區(qū)域的結(jié)構(gòu)預(yù)測(cè), 采用等變圖神經(jīng)網(wǎng)絡(luò)(E(n)-EGNNs)直接建模骨架原子的三維坐標(biāo), 能高效預(yù)測(cè)六個(gè)CDR環(huán)的結(jié)構(gòu), 尤其在CDR-H3這一結(jié)構(gòu)多樣性最高的區(qū)域表現(xiàn)突出. 與傳統(tǒng)基于模板的方法相比, ABlooper無(wú)需MSA, 在精度與速度上均具有優(yōu)勢(shì), 是一種高效���、實(shí)用的抗體結(jié)構(gòu)建模工具.IgFold [24]是近年來(lái)抗體結(jié)構(gòu)建模方法中的代表性工具之一, 它利用預(yù)訓(xùn)練抗體語(yǔ)言模型AntiBERTy進(jìn)行序列特征編碼, 并結(jié)合圖神經(jīng)網(wǎng)絡(luò)(graph neural networks,GNN)與不變點(diǎn)注意力(invariant point attention, IPA)模塊, 在無(wú)需MSA的情況下, 實(shí)現(xiàn)對(duì)輕鏈、重鏈及各CDR區(qū)域的高效建模. 與DeepAb等方法相比, IgFold在納米抗體結(jié)構(gòu)預(yù)測(cè)中整體表現(xiàn)優(yōu)良(CDR3平均精度接近AlphaFold), 且推理速度明顯更快; 需要說(shuō)明的是,ABlooper不支持納米抗體的結(jié)構(gòu)預(yù)測(cè), 因此未被納入納米抗體的對(duì)比. 依托這一速度優(yōu)勢(shì), IgFold更適合大規(guī)模���、高通量的抗體結(jié)構(gòu)建模. 具體見表3.
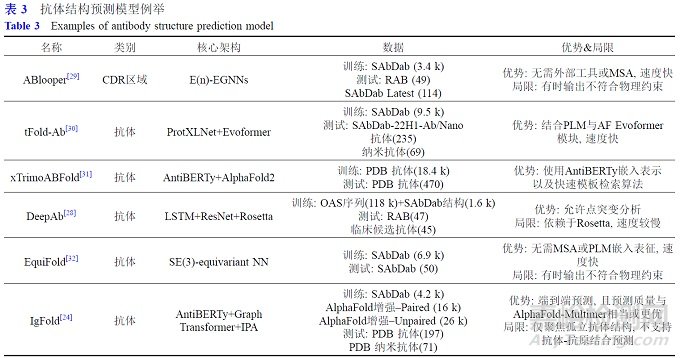
這些AI驅(qū)動(dòng)的抗體結(jié)構(gòu)預(yù)測(cè)方法共同構(gòu)建了一個(gè)高效����、精準(zhǔn)、面向功能的抗體建模體系. 它們顯著提升了對(duì)關(guān)鍵結(jié)構(gòu)區(qū)域, 特別是高變異性的CDR-H3環(huán)的建模能力, 同時(shí)加速了從序列到三維結(jié)構(gòu)的預(yù)測(cè)流程, 推動(dòng)抗體結(jié)構(gòu)設(shè)計(jì)與序列優(yōu)化的深度融合, 為從頭設(shè)計(jì)功能導(dǎo)向的新型抗體藥物提供了堅(jiān)實(shí)的技術(shù)支撐.
2.3 抗原-抗體相互作用預(yù)測(cè)
抗原-抗體相互作用是抗體發(fā)揮功能的基礎(chǔ), 因此抗原-抗體識(shí)別是決定抗體療效����、親和力和特異性的核心環(huán)節(jié), 其界面區(qū)域的精確建模直接影響后續(xù)的親和力成熟、免疫原性評(píng)估與結(jié)構(gòu)優(yōu)化策略. 隨著AI技術(shù)的不斷進(jìn)步, 抗原-抗體相互作用預(yù)測(cè)逐步從傳統(tǒng)的結(jié)構(gòu)對(duì)接方法轉(zhuǎn)向以GNN和蛋白質(zhì)表面幾何特征為核心的精準(zhǔn)界面建模路徑. PECAN [33]是一款面向抗體-抗原結(jié)合界面預(yù)測(cè)的模型, 它采用對(duì)稱圖卷積網(wǎng)絡(luò)(symmetric GCN), 將抗體與抗原的界面同時(shí)嵌入到網(wǎng)絡(luò)結(jié)構(gòu)中, 并顯式地引入另一方的信息作為上下文, 從而實(shí)現(xiàn)了抗原表位和抗體互補(bǔ)位的聯(lián)合�、協(xié)同預(yù)測(cè).盡管PECAN未在設(shè)計(jì)中顯式區(qū)分它們的功能角色或結(jié)構(gòu)特性, 但是模型在表位結(jié)合區(qū)預(yù)測(cè)中的PR-AUC達(dá)到0.70, 展示出良好的泛化能力. PECAN的設(shè)計(jì)強(qiáng)調(diào)跨界面互作建模, 使其適用于識(shí)別結(jié)合熱點(diǎn)、評(píng)估免疫結(jié)合位點(diǎn), 以及輔助抗體人源化和親和力優(yōu)化等下游任務(wù). 然而, 由于其依賴高質(zhì)量三維結(jié)構(gòu)作為輸入數(shù)據(jù), 模型在結(jié)構(gòu)不可獲得或存在構(gòu)象噪聲的場(chǎng)景下仍可能面臨性能瓶頸. 除了GNN對(duì)界面原子關(guān)系的建模路徑外, 幾何驅(qū)動(dòng)的分子表面建模方法也逐漸應(yīng)用于抗原-抗體識(shí)別問(wèn)題, 尤其擅長(zhǎng)捕捉結(jié)合界面的空間構(gòu)型與理化特性. MaSIF [34]是該類方法的代表模型之一, 最初用于蛋白-蛋白相互作用預(yù)測(cè), 它通過(guò)網(wǎng)格化重建的分子表面, 提取多尺度幾何與化學(xué)特征, 在界面識(shí)別任務(wù)中表現(xiàn)出色. 基于這一理念, 后續(xù)發(fā)展出的dMaSIF [35]對(duì)模型架構(gòu)進(jìn)行高效重構(gòu), 摒棄原有的網(wǎng)格重建流程, 直接從原子坐標(biāo)生成分子表面, 大幅提升了幾何特征提取速度(提升約600倍), 并在界面預(yù)測(cè)中取得ROC-AUC達(dá)到0.87的表現(xiàn). dMaSIF的輕量化設(shè)計(jì)特別適合大規(guī)?���?贵w-抗原表面識(shí)別任務(wù), 可支持實(shí)時(shí)交互界面搜索與高通量候選抗體評(píng)估, 在抗體工程和結(jié)構(gòu)設(shè)計(jì)流程中展現(xiàn)出良好的泛化能力和部署潛力. 相比PECAN和MaSIF依賴結(jié)構(gòu)輸入進(jìn)行建模的方法,AbAdapt [36]提供了一種從序列出發(fā)的替代方案, 適用于結(jié)構(gòu)不可獲得或信息缺失的場(chǎng)景. 該模型基于DNN的方法, 以抗體和抗原的序列為輸入, 配合結(jié)構(gòu)生成與分子對(duì)接流程, 用于識(shí)別潛在的結(jié)合界面. 盡管其在預(yù)測(cè)精度和運(yùn)行速度略低于幾何驅(qū)動(dòng)模型, 但在早期篩選或大規(guī)模評(píng)估等結(jié)構(gòu)不可獲得的場(chǎng)景中具有獨(dú)特優(yōu)勢(shì), 是連接序列數(shù)據(jù)與結(jié)構(gòu)建模的重要橋梁型工具. 具體見表4.
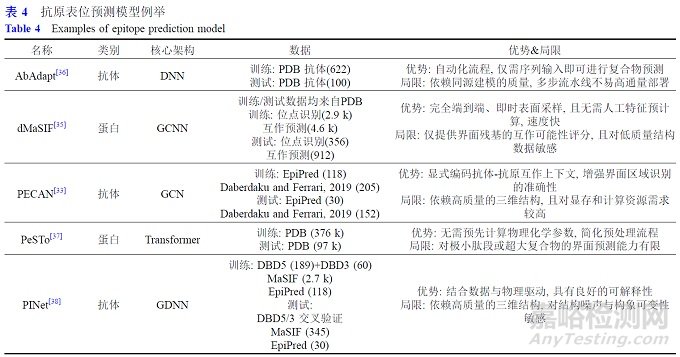
這些模型分別從圖結(jié)構(gòu)建模、表面幾何學(xué)習(xí)與序列驅(qū)動(dòng)生成等路徑切入, 通過(guò)引入上下文感知機(jī)制與多層次特征編碼, 顯著提升了抗原-抗體結(jié)合界面的識(shí)別與預(yù)測(cè)能力. 它們不僅增強(qiáng)了對(duì)互補(bǔ)位-表位互作模式的建模精度, 也拓展了在親和力優(yōu)化�、免疫原性評(píng)估及大規(guī)模抗體篩選中的應(yīng)用邊界, 正在成為AI驅(qū)動(dòng)抗體設(shè)計(jì)流程中的關(guān)鍵支撐模塊.
2.4 抗體親和力優(yōu)化
抗體親和力成熟是指抗體在免疫過(guò)程中通過(guò)體細(xì)胞高頻突變和選擇, 逐漸提高與抗原結(jié)合親和力的過(guò)程. 親和力成熟的抗體具有更高的特異性和親和力,能夠更有效地發(fā)揮免疫防御和治療作用. 因此, 提高抗體親和力成熟效率對(duì)于開發(fā)高效抗體藥物具有重要意義. 在抗體親和力優(yōu)化領(lǐng)域, AI技術(shù)使其從傳統(tǒng)依賴試錯(cuò)實(shí)驗(yàn)的模式, 逐步過(guò)渡到以序列數(shù)據(jù)驅(qū)動(dòng)的精準(zhǔn)預(yù)測(cè)與結(jié)構(gòu)生成相結(jié)合的高效優(yōu)化路徑. 這一過(guò)程本質(zhì)上是通過(guò)抗體序列的定向突變與篩選, 優(yōu)化其與特定抗原的結(jié)合能力, 從而顯著提升治療效果. 近年來(lái),研究者利用深度學(xué)習(xí)模型對(duì)大規(guī)??贵w-抗原結(jié)合數(shù)據(jù)進(jìn)行訓(xùn)練, 從而預(yù)測(cè)特定突變對(duì)親和力的提升潛力.例如, 最新提出的AffinityFlow [39]模型, 利用AlphaFold衍生的AlphaFlow模塊從序列預(yù)測(cè)抗原-抗體復(fù)合物結(jié)構(gòu), 并通過(guò)反向折疊(inverse folding)迭代優(yōu)化序列結(jié)構(gòu), 進(jìn)一步引入共教學(xué)(co-teaching)機(jī)制交替優(yōu)化. 先讓候選序列在復(fù)合體結(jié)構(gòu)上朝更有利的結(jié)合構(gòu)型收斂, 再據(jù)此篩選更可能提升親和力的突變. 公開的insilico測(cè)評(píng)顯示, AffinityFlow在CDR-H1/H2/H3及全環(huán)設(shè)計(jì)任務(wù)中, 其在功能性、特異性與自然度三類代理指標(biāo)上取得整體更均衡的表現(xiàn). 此外, 消融實(shí)驗(yàn)的結(jié)果表明: 去掉AlphaFlow引導(dǎo)���、能量約束或樣本選擇中的任何一環(huán), 效果都會(huì)有明顯回落; 完整流程則表現(xiàn)更穩(wěn)定. 與主要用于突變候選篩選的預(yù)測(cè)型模型不同, 生成導(dǎo)向的方法也開始用于重建親和力成熟過(guò)程.FvHallucinator [40]和RFdiffusion-Antibody [41]等結(jié)構(gòu)主導(dǎo)的模型, 基于目標(biāo)抗原-抗體結(jié)合界面的三維結(jié)構(gòu),采用“逆向設(shè)計(jì)”的策略, 在已有結(jié)合位點(diǎn)附近構(gòu)造出新的候選序列, 隨后借助結(jié)構(gòu)優(yōu)化流程進(jìn)行多輪循環(huán),篩選出符合穩(wěn)定折疊與高親和力要求的最優(yōu)序列. 此外, 基于語(yǔ)言模型的局部重構(gòu)方法在親和力優(yōu)化中同樣展現(xiàn)出獨(dú)特優(yōu)勢(shì). 不同于通過(guò)全局突變篩選或全序列生成的策略, 這類方法在保持抗體框架區(qū)穩(wěn)定的前提下, 針對(duì)CDR區(qū)域執(zhí)行上下文感知的“infill”式序列插入, 從而實(shí)現(xiàn)對(duì)關(guān)鍵互作位點(diǎn)的精細(xì)微調(diào). 進(jìn)一步地, 為避免出現(xiàn)“親和力最優(yōu)而可開發(fā)性不足”的情況,建議在上述生成-篩選流程之后, 引入面向成藥性的多目標(biāo)約束. 具體做法是, 以IgFold [24]或AlphaFold [13,27]得到的候選三維結(jié)構(gòu)作為統(tǒng)一評(píng)估輸入, 對(duì)入圍突變體同時(shí)開展三類評(píng)價(jià): 其一, 穩(wěn)定性與可折疊性, 采用Rosetta [42], FoldX [43]或DeepDDG [44]的ΔΔG估計(jì), 并輔以短程分子動(dòng)力學(xué)復(fù)核, 用以檢視界面能與幾何一致性; 其二, 免疫原性, 以MHC-II結(jié)合預(yù)測(cè)為主, 借助NetMHCIIpan [45]或IEDB [46]對(duì)CDR及新暴露表面進(jìn)行覆蓋式掃描; 其三, 溶解性�、黏度與聚集傾向, 依據(jù)序列與結(jié)構(gòu)特征給出快速風(fēng)險(xiǎn)評(píng)分 [47,48]. 隨后采用“兩階段”篩選: 先以輕量化代理指標(biāo)剔除高風(fēng)險(xiǎn)候選, 再對(duì)保留集合進(jìn)行精細(xì)重打分, 形成多屬性評(píng)分向量. 最后, 利用多目標(biāo)優(yōu)化篩選出“親和力-穩(wěn)定性-免疫原性-可開發(fā)性”的最優(yōu)解集, 并以小規(guī)模的BLI/SPR [49],ELISA以及SEC-HPLC/DLS [50]實(shí)驗(yàn)驗(yàn)證, 將結(jié)果用于持續(xù)校準(zhǔn)各代理模型與搜索權(quán)重 [51]. 具體見表5.
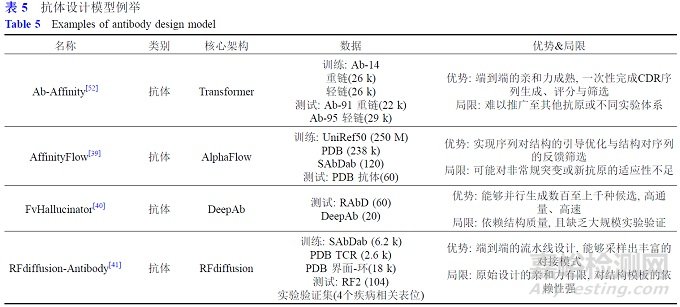
隨著這些AI技術(shù)在預(yù)測(cè)、結(jié)構(gòu)生成與序列微調(diào)各環(huán)節(jié)的深度融合, 抗體親和力優(yōu)化正朝著“算法-實(shí)驗(yàn)”閉環(huán)的智能化方向演進(jìn). AI不僅能夠從海量數(shù)據(jù)中精確識(shí)別最有潛力的突變位點(diǎn), 也可借助結(jié)構(gòu)與語(yǔ)言模型同步生成并打磨高親和力序列, 甚至通過(guò)貝葉斯優(yōu)化等策略動(dòng)態(tài)調(diào)整實(shí)驗(yàn)設(shè)計(jì)參數(shù), 如突變率與選擇壓力. 未來(lái), 隨著模型精度和數(shù)據(jù)規(guī)模的不斷提升, AI將日益成為親和力成熟流程中不可或缺的決策引擎, 為高效�、可控的抗體藥物開發(fā)提供堅(jiān)實(shí)支撐.
3 挑戰(zhàn)與機(jī)遇
近年來(lái), 隨著AI技術(shù)的飛速發(fā)展, 其在抗體藥物研發(fā)中的應(yīng)用日益廣泛, 為該領(lǐng)域帶來(lái)了前所未有的發(fā)展機(jī)遇. 借助大規(guī)模預(yù)訓(xùn)練語(yǔ)言模型����、結(jié)構(gòu)生成網(wǎng)絡(luò)與進(jìn)化算法, AI可顯著縮短抗體從初始篩選到優(yōu)化成藥的周期, 在疫情應(yīng)急、罕見病抗體藥物開發(fā)等場(chǎng)景中具備獨(dú)特價(jià)值. 在公共衛(wèi)生領(lǐng)域, AI通過(guò)從頭設(shè)計(jì)和對(duì)現(xiàn)有抗體的親和力成熟及抗逃逸能力的優(yōu)化, 快速生成突發(fā)傳染病抗體序列, 有效縮短研發(fā)周期并降低成本, 從加速響應(yīng)���、對(duì)抗變異到攻克難成藥靶點(diǎn)等方面為應(yīng)對(duì)突發(fā)公共衛(wèi)生事件以及病毒變異條件下抗體藥物的加速研發(fā)提供核心支撐. 更重要的是, AI將驅(qū)動(dòng)多特異性抗體�、納米抗體、抗體偶聯(lián)藥物等新型抗體藥物的快速設(shè)計(jì), 實(shí)現(xiàn)對(duì)復(fù)雜靶點(diǎn)或多重通路的精準(zhǔn)調(diào)控. 在精準(zhǔn)醫(yī)學(xué)背景下, AI還有望根據(jù)個(gè)體患者的免疫表型�、突變譜或病毒抗原表位變異情況, 定制個(gè)性化的抗體設(shè)計(jì)方案. 此外, AI方法的通用性也使其具備擴(kuò)展?jié)摿? 可應(yīng)用于如嵌合抗原受體(CAR)���、合成轉(zhuǎn)錄因子、酶工程����、新生抗原等非抗體蛋白的設(shè)計(jì)中, 助力合成生物學(xué)和個(gè)體化療法的技術(shù)發(fā)展.
盡管AI技術(shù)在抗體藥物設(shè)計(jì)領(lǐng)域取得了顯著進(jìn)展, 但要實(shí)現(xiàn)其在生物醫(yī)學(xué)中的全面應(yīng)用, 仍面臨一系列關(guān)鍵挑戰(zhàn). 首先, 當(dāng)前高質(zhì)量�、結(jié)構(gòu)解析明確的抗原-抗體復(fù)合體數(shù)據(jù)庫(kù)數(shù)量有限, 這在很大程度上限制了監(jiān)督學(xué)習(xí)模型的訓(xùn)練效果與泛化能力. 盡管如SAbDab[53], PDB [54]等數(shù)據(jù)庫(kù)持續(xù)更新, 但針對(duì)復(fù)雜構(gòu)象變異區(qū)域(如CDR-H3)或罕見抗原類型的數(shù)據(jù)依然稀缺.其次, 現(xiàn)有的深度學(xué)習(xí)模型多為“黑盒”結(jié)構(gòu), 缺乏對(duì)序列突變?nèi)绾我l(fā)結(jié)構(gòu)變化, 以及如何影響功能的解釋能力, 這在一定程度上限制了AI模型在抗體藥物研發(fā)中的應(yīng)用和信任度. 為提升可解釋性與可用性, 可在模型外加兩類路徑: 其一, 注意力權(quán)重-結(jié)構(gòu)/功能關(guān)聯(lián). AntiBERTy和IgLM等模型的注意力熱區(qū)映射到Fv三維結(jié)構(gòu)與CDR-H3關(guān)鍵殘基 [14,22,24,55]; 其二, 特征歸因算法——采用SHAP/Integrated Gradients對(duì)位點(diǎn)�、片段或界面補(bǔ)丁進(jìn)行貢獻(xiàn)度分解, 給出“哪一個(gè)突變���、為何有效”的可解釋證據(jù) [56,57]. 此外, 蛋白結(jié)構(gòu)預(yù)測(cè)、抗體-抗原對(duì)接和生成模型訓(xùn)練等過(guò)程均依賴于大規(guī)模高性能計(jì)算資源, 同時(shí)缺乏既快速又準(zhǔn)確的方法來(lái)評(píng)估AI模型生成的抗體效果, 導(dǎo)致驗(yàn)證成本增加, 制約了其在資源受限環(huán)境中的推廣. 最后, 目前的抗體成藥性優(yōu)化AI模型主要側(cè)重于優(yōu)化抗體結(jié)合親和力, 忽略了穩(wěn)定性和免疫原性等其他關(guān)鍵性質(zhì)的優(yōu)化. 抗體藥物設(shè)計(jì)本質(zhì)上是一個(gè)跨學(xué)科領(lǐng)域, AI模型若無(wú)法有效整合免疫學(xué)�、結(jié)構(gòu)生物學(xué)與計(jì)算建模之間的知識(shí),將難以實(shí)現(xiàn)面向?qū)嶋H應(yīng)用場(chǎng)景的高性能預(yù)測(cè)與生成.
未來(lái), AI驅(qū)動(dòng)的抗體設(shè)計(jì)將在模型體系�、設(shè)計(jì)流程和應(yīng)用場(chǎng)景等方面持續(xù)演進(jìn). 首先, 多模態(tài)學(xué)習(xí)框架將成為研究重點(diǎn), 通過(guò)融合蛋白序列���、三維結(jié)構(gòu)、分子動(dòng)力學(xué)模擬�、抗原表位信息以及實(shí)驗(yàn)驗(yàn)證數(shù)據(jù),建立更具全面性和魯棒性的集成預(yù)測(cè)系統(tǒng). 例如, 將AlphaFold結(jié)構(gòu)預(yù)測(cè)結(jié)果與分子表面潛能(如dMaSIF)相結(jié)合, 有望提升抗原-抗體結(jié)合位點(diǎn)預(yù)測(cè)的準(zhǔn)確性.其次, 抗體設(shè)計(jì)將邁向人機(jī)協(xié)同的閉環(huán)系統(tǒng): AI模型負(fù)責(zé)初步篩選和構(gòu)建候選序列, 實(shí)驗(yàn)人員快速完成表達(dá)與親和驗(yàn)證, 反饋數(shù)據(jù)再次訓(xùn)練優(yōu)化模型, 最終形成“AI-實(shí)驗(yàn)”互補(bǔ)增強(qiáng)的高通量抗體發(fā)現(xiàn)平臺(tái). 在可解釋AI方面, 未來(lái)的抗體建模不僅追求預(yù)測(cè)準(zhǔn)確率, 也更關(guān)注模型內(nèi)部機(jī)制與生物學(xué)可解釋性. 例如, 開發(fā)具備“結(jié)構(gòu)-功能”對(duì)齊機(jī)制的注意力網(wǎng)絡(luò), 使模型能夠明確指出某一突變?nèi)绾斡绊懹H和力����、熱穩(wěn)定性或免疫逃逸潛力, 從而為臨床設(shè)計(jì)和機(jī)制研究提供支持. 同時(shí), 新一代生成模型, 如擴(kuò)散模型���、圖生成網(wǎng)絡(luò)也已開始用于抗體CDR區(qū)域的設(shè)計(jì)與優(yōu)化, 能夠捕捉更復(fù)雜的結(jié)構(gòu)變異模式并生成具備生物可行性的候選序列. 最后,AI與合成生物學(xué)的融合將推動(dòng)智能免疫細(xì)胞(如CART/NK)、工程化抗體庫(kù)����、可控表達(dá)系統(tǒng)等平臺(tái)的發(fā)展,加速?gòu)膇n silico設(shè)計(jì)到in vivo驗(yàn)證的轉(zhuǎn)化效率, 開啟自動(dòng)化���、模塊化的抗體工程新范式.
4 結(jié)語(yǔ)
AI技術(shù)正在為抗體藥物設(shè)計(jì)帶來(lái)深刻變革, 重塑傳統(tǒng)抗體工程的流程與邊界. 從序列聚類、功能預(yù)測(cè)到三維結(jié)構(gòu)建模與抗原結(jié)合分析, AI模型實(shí)現(xiàn)了抗體設(shè)計(jì)的系統(tǒng)化與自動(dòng)化, 大幅提升了效率與精度. 特別是在親和力優(yōu)化�、多靶點(diǎn)識(shí)別和變異抗原適應(yīng)方面,AI展現(xiàn)出傳統(tǒng)方法難以比擬的優(yōu)勢(shì). 雖然當(dāng)前仍面臨數(shù)據(jù)匱乏、模型可解釋性差�、跨學(xué)科協(xié)同困難等挑戰(zhàn),但隨著算法優(yōu)化���、數(shù)據(jù)庫(kù)建設(shè)以及實(shí)驗(yàn)反饋機(jī)制的逐步完善, AI將成為推動(dòng)抗體藥物開發(fā)、疫苗設(shè)計(jì)與免疫治療升級(jí)的重要引擎. 未來(lái), AI驅(qū)動(dòng)的抗體工程將不僅服務(wù)于實(shí)驗(yàn)室研究, 更將在精準(zhǔn)醫(yī)療���、應(yīng)對(duì)突發(fā)傳染病和下一代生物治療中發(fā)揮不可替代的核心作用.
參考文獻(xiàn)
1 Meng Y, Zhang Z, Zhou C, et al. Protein structure prediction via deep learning: an in-depth review. Front Pharmacol, 2025, 16: 1498662
2 Jänes J, Beltrao P. Deep learning for protein structure prediction and design-progress and applications. Mol Syst Biol, 2024, 20: 162–169
3 Akdel M, Pires D E V, Pardo E P, et al. A structural biology community assessment of AlphaFold2 applications. Nat Struct Mol Biol, 2022, 29:1056–1067
4 Cheng J, Liang T, Xie X Q, et al. A new era of antibody discovery: an in-depth review of AI-driven approaches. Drug Discov Today, 2024, 29:103984
5 Kyro G W, Qiu T, Batista V S. A model-centric review of deep learning for protein design. arXiv, 2025, 2502.19173
6 Bai G, Sun C, Guo Z, et al. Accelerating antibody discovery and design with artificial intelligence: recent advances and prospects. Semin Cancer
Biol, 2023, 95: 13–24
7 Zheng J, Wang Y, Liang Q, et al. The application of machine learning on antibody discovery and optimization. Molecules, 2024, 29: 5923
8 LeCun Y, Boser B, Denker J S, et al. Backpropagation applied to handwritten zip code recognition. Neural Comput, 1989, 1: 541–551
9 Han Y, Kim D. Deep convolutional neural networks for pan-specific peptide-MHC class I binding prediction. BMC Bioinf, 2017, 18: 585
10 Hopfield J J. Neural networks and physical systems with emergent collective computational abilities. Proc Natl Acad Sci USA, 1982, 79: 2554–2558
11 Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput, 1997, 9: 1735–1780
12 Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. In Proceedings of the 31st International Conference on Neural InformationProcessing Systems (NIPS’17). NewYork: Curran Associates Inc., 2017. 6000–6010
13 Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature, 2021, 596: 583–589
14 Ruffolo J A, Gray J J, Sulam J. Deciphering antibody affinity maturation with language models and weakly supervised learning. arXiv, 2021,2112.07782
15 Fournier Q, Caron G M, Aloise D. A practical survey on faster and lighter transformers. ACM Comput Surv, 2023, 55: 1–40
16 Tay Y, Dehghani M, Bahri D, et al. Efficient transformers: a survey. ACM Comput Surv, 2023, 55: 1–28
17 Jain S, Wallace B C. Attention is not explanation. arXiv, 2019, 1902.10186
18 Evans R, O’Neill M, Pritzel A, et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv, 2021, 10.04.463034
19 Ho J, Jain A, Abbeel P. Denoising diffusion probabilistic models. In Proceedings of the 34th International Conference on Neural InformationProcessing Systems (NIPS’20). NewYork: Curran Associates Inc., 2020. 6840–6851
20 Hadsund J T, Sat?awa T, Janusz B, et al. nanoBERT: a deep learning model for gene agnostic navigation of the nanobody mutational space. BioinfAdv, 2024, 4: vbae033
21 Deszyński P, M?okosiewicz J, Volanakis A, et al. INDI-integrated nanobody database for immunoinformatics. Nucleic Acids Res, 2022, 50:D1273–D1281
22 Shuai R W, Ruffolo J A, Gray J J. IgLM: infilling language modeling for antibody sequence design. Cell Syst, 2023, 14: 979–989.e4
23 Olsen T H, Moal I H, Deane C M, et al. AbLang: an antibody language model for completing antibody sequences. Bioinf Adv, 2022, 2: vbac046
24 Ruffolo J A, Chu L S, Mahajan S P, et al. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. NatCommun, 2023, 14: 2389
25 Lin Z, Akin H, Rao R, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science, 2023, 379: 1123–1130
26 Kalemati M, Noroozi A, Shahbakhsh A, et al. ParaAntiProt provides paratope prediction using antibody and protein language models. Sci Rep,2024, 14: 29141
27 Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024, 630: 493–500
28 Ruffolo J A, Sulam J, Gray J J. Antibody structure prediction using interpretable deep learning. Patterns, 2022, 3: 100406
29 Abanades B, Georges G, Bujotzek A, et al. ABlooper: fast accurate antibody CDR loop structure prediction with accuracy estimation.Bioinformatics, 2022, 38: 1877–1880
30 Wu J, Wu F, Jiang B, et al. tFold-Ab: fast and accurate antibody structure prediction without sequence homologs. bioRxiv, 2022, 515918
31 Wang Y, Gong X, Li S, et al. xTrimoABFold: De novo antibody structure prediction without MSA. arXiv, 2022, 2212.00735
32 Kmiecik S, Gront D, Kolinski M, et al. Coarse-grained structure representation. bioRxiv, 2023, 511322
33 Pittala S, Bailey-Kellogg C. Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics,2020, 36: 3996–4003
34 Gainza P, Sverrisson F, Monti F, et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. NatMethods, 2020, 17: 184–192
35 Sverrisson F, Feydy J, Correia B E, et al. Fast End-to-End Learning on Protein Surfaces. NewYork: IEEE, 2021. 15272–15281
36 Davila A, Xu Z, Li S, et al. AbAdapt: an adaptive approach to predicting antibody-antigen complex structures from sequence. BioInf Adv, 2022,2: vbac015
37 Krapp L F, Abriata L A, Cortés Rodriguez F, et al. PeSTo: parameter-free geometric deep learning for accurate prediction of protein bindinginterfaces. Nat Commun, 2023, 14: 2175
38 Dai B, Bailey-Kellogg C, Ponty Y. Protein interaction interface region prediction by geometric deep learning. Bioinformatics, 2021, 37: 2580–2588
39 Chen C, Herpoldt K L, Zhao C, et al. AffinityFlow: guided flows for antibody affinity maturation. arXiv, 2025, 2502.10365
40 Mahajan S P, Ruffolo J A, Frick R, et al. Hallucinating structure-conditioned antibody libraries for target-specific binders. Front Immunol, 2022,13: 999034
41 Bennett N R, Watson J L, Ragotte R J, et al. Atomically accurate de novo design of antibodies with RFdiffusion. bioRxiv, 2025, 585103
42 Barlow K A, Ó Conchúir S, Thompson S, et al. Flex ddG: rosetta ensemble-based estimation of changes in protein-protein binding affinity uponmutation. J Phys Chem B, 2018, 122: 5389–5399
43 Schymkowitz J, Borg J, Stricher F, et al. The FoldX web server: an online force field. Nucleic Acids Res, 2005, 33: W382–W388
44 Cao H, Wang J, He L, et al. DeepDDG: predicting the stability change of protein point mutations using neural networks. J Chem Inf Model, 2019,59: 1508–1514
45 Reynisson B, Alvarez B, Paul S, et al. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrentmotif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res, 2020, 48: W449–W454
46 Kim Y, Ponomarenko J, Zhu Z, et al. Immune epitope database analysis resource. Nucleic Acids Res, 2012, 40: W525–W530
47 Zhang W, Wang H, Feng N, et al. Developability assessment at early-stage discovery to enable development of antibody-derived therapeutics.Antibody Ther, 2023, 6: 13–29
48 Chennamsetty N, Voynov V, Kayser V, et al. Prediction of aggregation prone regions of therapeutic proteins. J Phys Chem B, 2010, 114: 6614–6624
49 Murali S, Rustandi R, Zheng X, et al. Applications of surface plasmon resonance and biolayer interferometry for virus-ligand binding. Viruses,2022, 14: 717
50 Alhazmi H A, Albratty M. Analytical techniques for the characterization and quantification of monoclonal antibodies. Pharmaceuticals, 2023, 16:291
51 Narayanan H, Dingfelder F, Condado Morales I, et al. Design of biopharmaceutical formulations accelerated by machine learning. Mol Pharm2021, 18: 3843–3853
52 Bin Ashraf F, Paco K, Zhang Z, et al. A large language model guides the affinity maturation of antibodies generated by combinatorialoptimization algorithms. bioRxiv, 2025, 629473
53 Dunbar J, Krawczyk K, Leem J, et al. SAbDab: the structural antibody database. Nucleic Acids Res, 2014, 42: D1140–D1146
54 Berman H M. The protein data bank. Nucleic Acids Res, 2000, 28: 235–242
55 Leem J, Mitchell L S, Farmery J H R, et al. Deciphering the language of antibodies using self-supervised learning. Patterns, 2022, 3: 100513
56 Gelman S, Fahlberg S A, Heinzelman P, et al. Neural networks to learn protein sequence-function relationships from deep mutational scanningdata. Proc Natl Acad Sci USA, 2021, 118: e2104878118
57 Sun Y, Wang R, Luo Z, et al. ESM2_AMP: an interpretable framework for protein-protein interactions prediction and biological mechanismdiscovery. Brief BioInf, 2025, 26: bbaf434


