一����、研究標(biāo)題(Study Title)
1.法規(guī)要求
21 CFR Part 58僅要求標(biāo)題“具有描述性”(descriptive)����,但未給出具體標(biāo)準(zhǔn),表述較為模糊��。
2.行業(yè)最佳實(shí)踐
為便于識(shí)別����、歸檔和監(jiān)管審評(píng),建議采用標(biāo)準(zhǔn)化格式的標(biāo)題��,包含以下關(guān)鍵信息:
3.示例
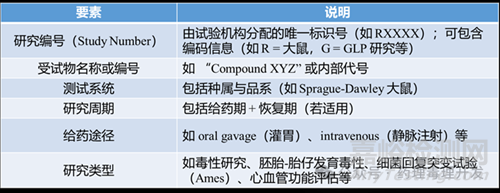
RXXXX:A 28-day oral gavage toxicity study of Compound XYZ in Sprague-Dawley Rats with a14-day recovery period.
(RXXXX:化合物XYZ在Sprague-Dawley大鼠中經(jīng)口灌胃給藥28天毒性研究����,含14天恢復(fù)期����。)
4.編號(hào)管理說(shuō)明
CRO(合同研究組織)通常根據(jù)其主計(jì)劃表(Master Schedule)分配純數(shù)字型唯一研究編號(hào)����。申辦方(Sponsor)常另設(shè)一套內(nèi)部研究編號(hào),用于自身數(shù)據(jù)管理和申報(bào)文件組織��,可能與CRO編號(hào)不同����。
標(biāo)準(zhǔn)化標(biāo)題有助于監(jiān)管人員快速把握研究設(shè)計(jì)、階段及用途����,提升審評(píng)效率�����。
二����、研究目的(Purpose of the Study)
1.定位與作用
是對(duì)研究核心目標(biāo)的簡(jiǎn)明陳述,屬于方案中的基本要素;可與標(biāo)題內(nèi)容高度重合�����,但應(yīng)更完整����、更具描述性。
2.撰寫建議
應(yīng)清晰說(shuō)明:研究終點(diǎn)(如毒性�����、毒代動(dòng)力學(xué))�����;動(dòng)物性別與種屬�����;給藥方案(途徑��、周期)����;是否包含恢復(fù)期評(píng)估�����。
3.示例
“To determine the toxicity and toxicokinetics of Compound X in male and female rats when administered by oral gavage for 28 days, and to evaluate recovery from any potential toxic effects.”
(旨在評(píng)估化合物X經(jīng)口灌胃給藥28天后在雄性和雌性大鼠中的毒性及毒代動(dòng)力學(xué)特征����,并考察潛在毒性效應(yīng)的可逆性�����。)
三����、受試物與對(duì)照物的識(shí)別(Identification of Test and Control Articles)
1. 基本識(shí)別信息
必須提供:名稱、化學(xué)文摘號(hào)(CAS No.)或內(nèi)部代碼編號(hào)����;
通常情況:受試物名稱多為企業(yè)內(nèi)部化合物代號(hào)(如“Compound XYZ”)。
2. 推薦補(bǔ)充信息(雖非強(qiáng)制����,但強(qiáng)烈建議)
目的:確保不同研究間數(shù)據(jù)可比性����,支持科學(xué)解讀與監(jiān)管評(píng)估��。
3.對(duì)照物與參比物(Reference Article)
OECD GLP明確要求識(shí)別參比物質(zhì)(Reference Item��,即對(duì)照物質(zhì))����;
FDA GLP(21 CFR Part 58)未明確定義“參比物”����,但實(shí)踐中通常包含陽(yáng)性/陰性對(duì)照或溶媒對(duì)照,并需明確標(biāo)識(shí)�����。
4.儲(chǔ)存條件與留樣要求
儲(chǔ)存條件
方案中應(yīng)明確受試物����、對(duì)照物及留樣(reserve samples)的儲(chǔ)存要求,包括:溫度范圍����、濕度控制、避光要求等��。
留樣(Reserve Samples)
定義:指研究中所用每一批次受試物的代表性樣品�����;
法規(guī)依據(jù):21 CFR 211.170(雖屬cGMP條款,但常被參考用于GLP研究)規(guī)定:1)留樣量:至少為完成全部質(zhì)量檢測(cè)所需量的兩倍(不包括無(wú)菌和熱原檢測(cè))��;2)保存期限:需在試驗(yàn)機(jī)構(gòu)保存至研究結(jié)束后規(guī)定時(shí)間(通常覆蓋整個(gè)研究周期及必要復(fù)測(cè)窗口)��。
5.關(guān)于批次號(hào)(Lot/Batch Number)的最佳實(shí)踐
是否強(qiáng)制����?否,非GLP法規(guī)硬性要求�����。
不過(guò)�����,推薦做法是在方案中注明所用受試物的生產(chǎn)批次號(hào)��。若研究中途更換批次��,需通過(guò)方案修訂(amendment)記錄����。
新舊批次使用日期需明確標(biāo)注,便于:1)分析治療期間出現(xiàn)的毒性是否與批次相關(guān)����;2)評(píng)估批次特異性雜質(zhì)的影響;3)雖然批次信息通常記錄在原始數(shù)據(jù)或最終報(bào)告中����,但寫入方案更便于快速查閱。
6.GMP生產(chǎn)的API能否用于GLP研究����?
1)法規(guī)立場(chǎng)
GLP研究不要求使用GMP物料。
但允許使用GMP生產(chǎn)的API��,前提是:在GLP研究報(bào)告中聲明此為“GLP例外”(GLP exception)����,因GMP與GLP在質(zhì)量體系上存在差異。提供完整的質(zhì)量分析證書(Certificate of Analysis, CoA)�����。
2)FDA接受情況
FDA已接受使用以下物料開展的非臨床研究:1)GMP生產(chǎn)的API(常見于為I期臨床制備的原料)��;2)非GMP/非GLP的工程批次(engineering batches)�����,前提是:API表征充分;CoA包含覆蓋整個(gè)給藥周期的有效期或復(fù)驗(yàn)期�����;生產(chǎn)商具備可靠�����、可重現(xiàn)的生產(chǎn)工藝記錄��。
3)風(fēng)險(xiǎn)提示
使用未充分表征的非GLP API存在監(jiān)管拒收風(fēng)險(xiǎn)����。若API純度��、雜質(zhì)譜或穩(wěn)定性不明�����,可能導(dǎo)致非臨床數(shù)據(jù)不被認(rèn)可��,影響后續(xù)臨床或注冊(cè)進(jìn)程。
四�����、非臨床研究方案中“申辦方”與“試驗(yàn)機(jī)構(gòu)”信息的填寫要求
1.基本內(nèi)容(FDA要求)
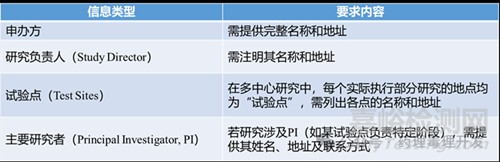
方案中需明確:申辦方(Sponsor)����;開展研究的試驗(yàn)機(jī)構(gòu)名稱和地址(Testing Facility)����。此項(xiàng)要求相對(duì)直接,但實(shí)際操作中需注意細(xì)節(jié)�����,尤其在多地點(diǎn)或跨國(guó)研究中��。
2.OECD GLP的更詳細(xì)規(guī)定
OECD GLP對(duì)相關(guān)信息提出了更全面的要求����,包括:
根據(jù)OECD定義:
“試驗(yàn)機(jī)構(gòu)”=研究負(fù)責(zé)人(SD)所在地點(diǎn);
“試驗(yàn)點(diǎn)”=PI所在的其他執(zhí)行地點(diǎn)��。
3.行業(yè)實(shí)踐與監(jiān)管考量
盡管FDA法規(guī)未強(qiáng)制要求提供所有地址和人員信息����,但目前全球申報(bào)實(shí)踐中����,方案普遍包含:申辦方����、試驗(yàn)機(jī)構(gòu)、各試驗(yàn)點(diǎn)��、研究負(fù)責(zé)人��、主要研究者的姓名��、地址及聯(lián)系方式��。這已成為國(guó)際監(jiān)管申報(bào)的標(biāo)準(zhǔn)格式��。
4.地址信息的重要性
許多CRO擁有多個(gè)分支機(jī)構(gòu)或?qū)嶒?yàn)室����。
明確標(biāo)注具體開展研究的地點(diǎn)地址至關(guān)重要,原因包括:1)若該地點(diǎn)曾被FDA發(fā)出483缺陷通知或存在合規(guī)問(wèn)題��,監(jiān)管機(jī)構(gòu)可快速核查����;2)在FDA審計(jì)或質(zhì)疑研究數(shù)據(jù)完整性時(shí)�����,能準(zhǔn)確定位責(zé)任單位��;3)有助于確保審計(jì)追蹤(audit trail)的準(zhǔn)確性和透明度����。
五����、非臨床試驗(yàn)系統(tǒng)(數(shù)量�����、體重��、性別��、來(lái)源�����、種屬、品系/亞系�����、年齡等)
1.測(cè)試系統(tǒng)的關(guān)鍵特征及其科學(xué)意義
在非臨床毒理學(xué)研究中��,必須在方案中明確以下試驗(yàn)系統(tǒng)特征(通常為實(shí)驗(yàn)動(dòng)物):
所有這些變量均可能影響受試物的藥代動(dòng)力學(xué)����、毒代動(dòng)力學(xué)及毒性表現(xiàn),是跨研究數(shù)據(jù)比較和風(fēng)險(xiǎn)評(píng)估的基礎(chǔ)����。
2.各要素的具體要求與最佳實(shí)踐
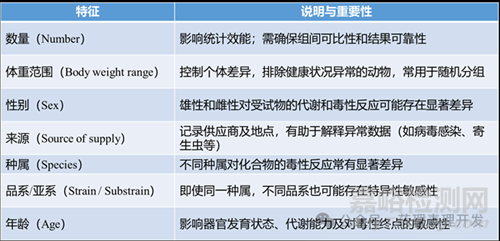
1)動(dòng)物數(shù)量(Group Size)
如一般亞慢性毒性研究(如28天、90天)����,嚙齒類通常每組每性別10–12只(行業(yè)標(biāo)準(zhǔn),雖非法規(guī)強(qiáng)制)����。當(dāng)然,國(guó)內(nèi)數(shù)量要求更多一些�����,一般不少于15只/性別。
生殖毒性研究(如胚胎-胎仔發(fā)育)要求16–20只孕鼠(ICH Draft Guideline, 2017)�����;致癌性研究通常需要更大樣本量以確保統(tǒng)計(jì)效力�����。
目標(biāo)是在倫理(3R原則)與科學(xué)可靠性之間取得平衡����。另外,國(guó)內(nèi)外對(duì)動(dòng)物數(shù)量要求可能與此不一致��,可參考具體指導(dǎo)原則����。
2)體重范圍
設(shè)定合理范圍:減少個(gè)體變異性��;識(shí)別并剔除健康狀況不佳的“離群值”動(dòng)物�����;作為隨機(jī)分組依據(jù)����,避免組間基線偏差����。
3)性別選擇
常規(guī)毒理研究:必須同時(shí)使用雄性和雌性動(dòng)物��;
例外情況:生殖毒性研究(如僅用孕雌性)����;安全藥理學(xué)研究(若已知無(wú)性別差異)。
性別差異的典型案例(嚙齒類):雄性大鼠細(xì)胞色素P450酶水平更高→藥物清除更快�����,可能導(dǎo)致系統(tǒng)暴露量(AUC)降低��。需采用不同劑量�����,使兩性暴露相當(dāng)����。
4)動(dòng)物來(lái)源(Supplier)
應(yīng)記錄供應(yīng)商名稱及設(shè)施地址;
原因:某些供應(yīng)商可能存在隱性感染(如病毒����、寄生蟲)����,影響藥物反應(yīng)��;遺傳漂變(Genetic drift)可能導(dǎo)致背景病理發(fā)生率變化����,生殖指標(biāo)波動(dòng)。
優(yōu)質(zhì)供應(yīng)商通常提供歷史繁殖與健康數(shù)據(jù)��,有助于數(shù)據(jù)解讀與跨研究比對(duì)�����。
5)種屬與品系
種屬差異:極為常見(如犬對(duì)某些心臟毒性更敏感�����,猴更接近人)����;
品系差異:較少但存在�����,例如:Brown Norway大鼠對(duì)藥物誘導(dǎo)的皮膚藥疹高度敏感。
選擇應(yīng)基于科學(xué)合理性與監(jiān)管接受度����。
6)年齡要求
比如一般毒理學(xué)研究,使用性成熟青年動(dòng)物為默認(rèn)標(biāo)準(zhǔn)����,如大鼠8-10周齡,犬5-7個(gè)月(國(guó)內(nèi)要求6-12月)��。而慢性/致癌性研究����,則需確保足夠動(dòng)物存活至研究結(jié)束,選擇更年幼動(dòng)物��,如大鼠5-6周齡�����。當(dāng)然����,幼齡動(dòng)物研究�����,則要根據(jù)目標(biāo)患者年齡��、靶器官毒性及給藥窗口確定�����。
如果使用未成熟動(dòng)物��,需謹(jǐn)慎評(píng)估其對(duì)發(fā)育中器官系統(tǒng)(如生殖����、神經(jīng)�����、免疫)的影響��。
3.試驗(yàn)系統(tǒng)的識(shí)別
1)個(gè)體識(shí)別的重要性
在非臨床毒理學(xué)研究中����,對(duì)每只實(shí)驗(yàn)動(dòng)物進(jìn)行準(zhǔn)確識(shí)別至關(guān)重要����,原因包括:確保正確動(dòng)物接受正確劑量��;保證各項(xiàng)操作(如采樣��、觀察�����、解剖)����;避免數(shù)據(jù)混淆����,保障數(shù)據(jù)完整性與可追溯性。
2)常用識(shí)別方法
主要識(shí)別方式(推薦用于GLP研究)為植入式微型芯片(Microchip):通過(guò)計(jì)算機(jī)連接的掃描儀讀取唯一編號(hào)��;可自動(dòng)記錄并追蹤動(dòng)物接受的各項(xiàng)操作����;是當(dāng)前GLP毒理實(shí)驗(yàn)室的主流做法。
輔助/備用識(shí)別方式:皮膚紋身(Tattoo)�����、耳標(biāo)、永久性記號(hào)筆標(biāo)記
這些手動(dòng)方法通常作為微芯片失效時(shí)的備份手段��,確保即使技術(shù)故障也能維持個(gè)體識(shí)別�����。
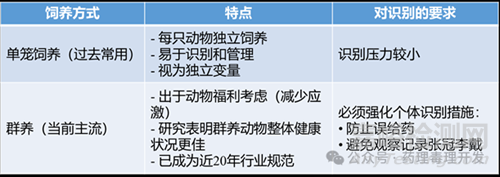
3)群養(yǎng)vs.單籠飼養(yǎng)對(duì)識(shí)別的影響
特別注意:某些個(gè)體(尤其是雄性小鼠)具有較強(qiáng)攻擊性��,易與籠伴打斗����。此類動(dòng)物需臨時(shí)或永久單籠飼養(yǎng),并在方案或原始記錄中說(shuō)明原因�����。
六��、試驗(yàn)設(shè)計(jì)描述及偏倚控制方法
1.試驗(yàn)設(shè)計(jì)描述
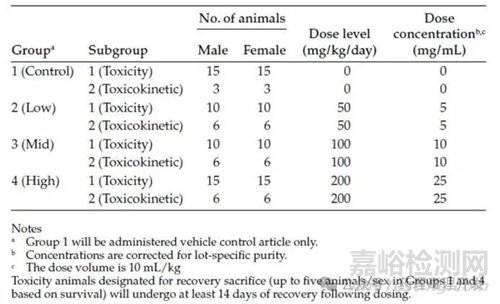
試驗(yàn)設(shè)計(jì)應(yīng)在研究方案中清晰說(shuō)明��,通常以表格形式呈現(xiàn)����,包含以下關(guān)鍵要素:研究分組(Study Groups)��,明確各組設(shè)置(如對(duì)照組��、低/中/高劑量組);每組動(dòng)物數(shù)量及性別分布�����,如“10只雄性+10只雌性/組”�����;劑量水平(Dose Levels)�����,以mg/kg或其他合適單位表示�����;給藥體積與濃度�����,確保給藥一致性(如5mL/kg��,10mg/mL);衛(wèi)星組(Satellite Groups)��,包括用于毒代動(dòng)力學(xué)(TK)或恢復(fù)期觀察的額外動(dòng)物����;對(duì)照組類型,標(biāo)注陰性對(duì)照(如溶媒對(duì)照)����、陽(yáng)性對(duì)照(如已知毒性化合物)。此類表格有助于審評(píng)人員快速理解整體研究架構(gòu)和劑量-反應(yīng)關(guān)系設(shè)計(jì)����。如下表所示。
其它如給藥途徑(對(duì)于靜脈輸注(IV infusion)研究��,需注明輸注速率(如 mg/kg/h)��、輸注持續(xù)時(shí)間(如 30 分鐘����、24 小時(shí)))、給藥頻率和周期也應(yīng)在方案中注明��。此外��,方案中還應(yīng)注明給藥途徑(如與臨床擬用途徑一致)、給藥劑量選擇的理由����。
2.偏倚控制方法(Control of Bias)
為確保數(shù)據(jù)客觀性和科學(xué)可靠性,需在關(guān)鍵環(huán)節(jié)采取系統(tǒng)性措施減少偏倚:
1)動(dòng)物隨機(jī)分組(Randomization)
嚙齒類動(dòng)物(大鼠��、小鼠等):采用計(jì)算機(jī)生成的隨機(jī)分配程序��;優(yōu)先使用能平衡各組體重分布的算法(如區(qū)組隨機(jī)化)����,避免因體重差異引入混雜因素�����。
大型動(dòng)物(犬��、非人靈長(zhǎng)類�����、小型豬等):隨機(jī)化前進(jìn)行給藥前篩選����,包括臨床病理學(xué)檢查(血液學(xué)����、生化等)��;心電圖(ECG)評(píng)估����。
排除標(biāo)準(zhǔn):存在明顯異常指標(biāo)的個(gè)體;無(wú)法適應(yīng)群養(yǎng)(如攻擊性強(qiáng))的動(dòng)物����。
2)解剖順序隨機(jī)化
在剖檢階段,按劑量組隨機(jī)或交錯(cuò)順序進(jìn)行操作�����,目的是避免技術(shù)人員因知曉劑量信息而無(wú)意識(shí)產(chǎn)生觀察或記錄偏差(如對(duì)高劑量組更仔細(xì)檢查)�����。
七����、非臨床GLP研究中飼料��、飲水及溶媒/助懸劑等輔料的描述要求與質(zhì)量控制規(guī)范
1.飼料、溶媒及其他輔料的描述要求
根據(jù)GLP法規(guī)(如21CFRPart58)��,研究方案中必須明確說(shuō)明以下內(nèi)容:
1)飼料信息
種類與來(lái)源:需注明所用飼料的名稱��、供應(yīng)商及是否為標(biāo)準(zhǔn)實(shí)驗(yàn)動(dòng)物飼料����;
污染物控制要求:必須提供飼料中可合理預(yù)期存在的污染物(如重金屬、農(nóng)藥殘留�����、霉菌毒素�����、植物雌激素等)����;
需設(shè)定可接受的限量標(biāo)準(zhǔn)(specifications)����,確保這些污染物不會(huì)干擾研究目的或結(jié)果。
特別注意:大豆類嚙齒類飼料中含有的植物雌激素(如染料木黃酮)可能影響發(fā)育毒性或內(nèi)分泌干擾研究的結(jié)果�����,因此在相關(guān)研究中應(yīng)謹(jǐn)慎選擇低雌激素或無(wú)大豆飼料。
2)溶媒/助懸劑/載體(Vehicle)
常用輔料包括:羥丙甲纖維素(HPMC)����、羧甲基纖維素(CMC)、吐溫80����、生理鹽水、植物油等����;
要求:使用公認(rèn)無(wú)毒、藥用級(jí)或分析純級(jí)別的材料��;提供制造商的純度分析報(bào)告(Certificate of Analysis, CoA)����;批次信息需記錄,并能與研究數(shù)據(jù)關(guān)聯(lián)�����。
3)混合前處理說(shuō)明
若受試物需通過(guò)溶劑溶解、乳化或懸浮后再與載體(如飼料�����、水��、灌胃液)混合��,應(yīng)詳細(xì)描述該過(guò)程�����。
包括:輔料名稱、濃度��、配制方法����、穩(wěn)定性數(shù)據(jù)(如適用)��。
2.飼料與飲水的質(zhì)量控制實(shí)踐
1)飼料
通常由大型實(shí)驗(yàn)動(dòng)物飼料生產(chǎn)商供應(yīng)��;每批飼料均進(jìn)行污染物檢測(cè)(如霉菌毒素、營(yíng)養(yǎng)成分、雌激素活性物質(zhì))����;檢測(cè)報(bào)告由試驗(yàn)機(jī)構(gòu)歸檔,并與具體研究關(guān)聯(lián)�����。
2)飲水
一般來(lái)源于市政飲用水系統(tǒng)�����,市政部門或第三方實(shí)驗(yàn)室定期提供水質(zhì)分析報(bào)告����。
試驗(yàn)機(jī)構(gòu)可額外檢測(cè):微生物(如細(xì)菌總數(shù)�����、大腸桿菌)�����;氯含量��、pH值、重金屬等����。
目的:確保動(dòng)物房供水無(wú)污染、適于實(shí)驗(yàn)動(dòng)物長(zhǎng)期飲用��。
飼料和飲水的質(zhì)量直接影響動(dòng)物健康狀態(tài)及藥物反應(yīng)�����,必須在方案中明確規(guī)格并持續(xù)監(jiān)控��。
3.GLP合規(guī)性要求:記錄審查與文檔留存
研究負(fù)責(zé)人(Study Director)和試驗(yàn)機(jī)構(gòu)管理層必須:定期審查飼料��、飲水����、溶媒等輔料的質(zhì)量檢測(cè)記錄;書面記錄審查過(guò)程及對(duì)研究潛在影響的評(píng)估��;將相關(guān)文件(如CoA��、水質(zhì)報(bào)告�����、內(nèi)部檢測(cè)結(jié)果)作為原始數(shù)據(jù)的一部分歸檔����。
FDA在GLP檢查中曾多次因以下問(wèn)題發(fā)出483缺陷項(xiàng):研究負(fù)責(zé)人未審查飼料/飲水污染物數(shù)據(jù);未記錄審查結(jié)果或影響評(píng)估��;無(wú)法證明輔料質(zhì)量不影響研究完整性��。
八�����、檢測(cè)�����、分析項(xiàng)目及其頻率的描述要求
1.核心內(nèi)容:觀察�����、評(píng)估與操作的時(shí)間安排
研究方案中必須明確說(shuō)明:將進(jìn)行哪些類型的檢測(cè)�����、分析和測(cè)量(如臨床觀察�����、血液學(xué)��、生化����、組織病理學(xué)、心電圖等)����;
每項(xiàng)操作的具體時(shí)間點(diǎn)和頻率(如給藥前、每周一次����、末次給藥后24小時(shí)、恢復(fù)期結(jié)束等)����。
這些內(nèi)容共同構(gòu)成研究的觀察計(jì)劃(Observation Schedule),是確保數(shù)據(jù)系統(tǒng)性和完整性的重要依據(jù)����。
2.操作細(xì)節(jié)的呈現(xiàn)方式
詳細(xì)操作步驟(如采血技術(shù)、麻醉方法����、解剖流程等)通常不重復(fù)寫入方案,而是引用試驗(yàn)機(jī)構(gòu)已有的標(biāo)準(zhǔn)操作規(guī)程(SOP)��;在方案中注明“按SOP編號(hào)XXX執(zhí)行”即可�����。
但以下與特定研究或評(píng)估相關(guān)的關(guān)鍵細(xì)節(jié)必須在方案中明確說(shuō)明:
這些細(xì)節(jié)直接影響樣本質(zhì)量和檢測(cè)結(jié)果可靠性�����,不能僅依賴SOP��,需在方案中具體規(guī)定����。
總之,建議使用表格形式列出所有檢測(cè)項(xiàng)目�����、時(shí)間點(diǎn)�����、頻率及樣本要求,提升可讀性����;對(duì)關(guān)鍵毒性終點(diǎn)(如神經(jīng)行為、生殖功能�����、免疫指標(biāo))的評(píng)估方法應(yīng)特別詳盡��;若某項(xiàng)檢測(cè)采用非標(biāo)準(zhǔn)方法或定制化流程��,必須在方案中完整描述����,不可僅引用SOP;所有引用的SOP應(yīng)為現(xiàn)行有效版本��,并在原始數(shù)據(jù)中可追溯�����。
九��、非臨床GLP研究中需保存的記錄類型及法定/行業(yè)保留期限要求
1.需歸檔保存的記錄范圍(依據(jù)21 CFR 58.190)
為確保研究可完整重建(reconstruct the study)��,以下材料必須歸檔保存:
所有電子與紙質(zhì)記錄原則上均應(yīng)歸檔,除非明確不屬于“原始數(shù)據(jù)”(參見法規(guī)對(duì)原始數(shù)據(jù)的定義)�����。
2.記錄保存期限(依據(jù)21 CFR 58.195)
保存期限取決于研究用途及申報(bào)狀態(tài)����,具體如下:
1)支持獲批上市申請(qǐng)的研究
適用情形:研究數(shù)據(jù)用于支持已獲FDA批準(zhǔn)的藥品/器械上市申請(qǐng)(NDA�����、BLA�����、PMA等)�����;
保存期限:自FDA批準(zhǔn)之日起�����,至少保存2年����。
2)支持IND或IDE的研究
適用情形:研究用于新藥臨床試驗(yàn)申請(qǐng)(IND)����;
保存期限:自向FDA提交該研究數(shù)據(jù)之日起��,至少保存5年��。
注意:盡管IND/IDE本身未獲“最終批準(zhǔn)”�����,但因其處于研發(fā)關(guān)鍵階段�����,監(jiān)管要求更長(zhǎng)的保存期�����。
3)未用于任何申報(bào)的研究
適用情形:研究完成后未提交給FDA用于任何申請(qǐng)(如早期篩選�����、終止項(xiàng)目);
保存期限:自研究完成��、終止或中止之日起��,至少保存2年����。
3.CRO實(shí)際操作慣例
多數(shù)CRO在協(xié)議中約定:研究結(jié)束后先由CRO保存1年;屆時(shí)聯(lián)系申辦方(Sponsor)����,確認(rèn)檔案后續(xù)處置方式(繼續(xù)保存����、轉(zhuǎn)移或銷毀);此做法便于申辦方根據(jù)項(xiàng)目進(jìn)展決定長(zhǎng)期歸檔策略�����。
4.企業(yè)額外保留策略(超出法規(guī)最低要求)
部分公司出于以下考慮����,選擇無(wú)限期保存(indefinite retention):藥物未來(lái)可能拓展新適應(yīng)癥;應(yīng)對(duì)潛在監(jiān)管問(wèn)詢或訴訟�����;支持生命周期管理或?qū)@m紛;此類決策通?����;趦?nèi)部質(zhì)量政策或風(fēng)險(xiǎn)管理框架��。
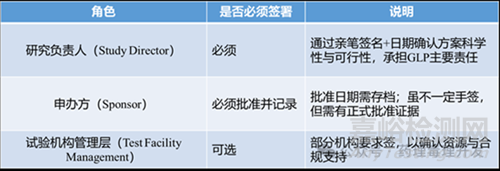
十�����、非臨床GLP研究中方案批準(zhǔn)的規(guī)范要求與各方職責(zé)
1.方案批準(zhǔn)的核心要求
根據(jù)GLP規(guī)范�����,研究方案批準(zhǔn)必須包含以下兩項(xiàng)關(guān)鍵信息:申辦方(Sponsor)�����;研究負(fù)責(zé)人(Study Director)�����。此過(guò)程也稱為“方案定稿”(protocol finalization)�����,是研究正式啟動(dòng)前的必要合規(guī)步驟。
2.簽署主體與責(zé)任分工
若使用電子簽名系統(tǒng)�����,需符合21 CFR Part 11等法規(guī)要求�����。
所有方案及后續(xù)修訂的申辦方批準(zhǔn)記錄����,必須保存在研究往來(lái)文件(study correspondence file)中����,供未來(lái)核查或?qū)徲?jì)使用。
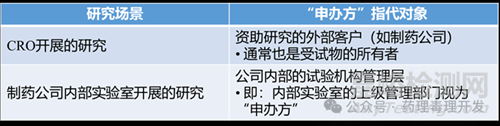
3.“申辦方”的定義因研究場(chǎng)所而異
可以參考OECD對(duì)申辦方與試驗(yàn)機(jī)構(gòu)的定義�����。
十一��、統(tǒng)計(jì)方法描述要求
1. 撰寫責(zé)任
統(tǒng)計(jì)方法部分應(yīng)由熟悉研究設(shè)計(jì)和所用統(tǒng)計(jì)軟件的專業(yè)統(tǒng)計(jì)人員撰寫?�,F(xiàn)代實(shí)驗(yàn)室數(shù)據(jù)系統(tǒng)通常內(nèi)置統(tǒng)計(jì)模塊或與專業(yè)統(tǒng)計(jì)軟件(如 SAS、R��、JMP)集成��。
2. 法規(guī)要求
GLP 法規(guī)(如 21 CFR Part 58或 OECD GLP)未強(qiáng)制規(guī)定具體統(tǒng)計(jì)方法��;方法選擇由試驗(yàn)機(jī)構(gòu)和研究負(fù)責(zé)人(Study Director)根據(jù)科學(xué)合理性決定����。
3. 常規(guī)實(shí)踐
所有成熟GLP實(shí)驗(yàn)室均配備統(tǒng)計(jì)支持人員;
最常用方法:方差分析(ANOVA)��;事后多重比較檢驗(yàn)(Post-hoc tests��,如 Dunnett’s test)����;以同期對(duì)照組(concurrent control)為基準(zhǔn),評(píng)估受試物處理效應(yīng)����。
目的:確保數(shù)據(jù)分析方法科學(xué)、透明����、可重復(fù)�����,并能有效識(shí)別劑量-反應(yīng)關(guān)系或毒性信號(hào)����。
十二��、方案修訂與偏離的管理規(guī)范
方案修訂(Amendment)與偏離(Deviation)的管理
1. 方案修訂(Protocol Amendment)
定義:指研究啟動(dòng)后(即研究負(fù)責(zé)人簽署方案之后)對(duì)原方案的有意�����、計(jì)劃性修改��。
最佳實(shí)踐:采用“重發(fā)完整方案 + 變更高亮”方式�����,提升可讀性與合規(guī)性��。
2. 方案偏離(Protocol Deviation)
監(jiān)管風(fēng)險(xiǎn)
FDA 在檢查中常因以下問(wèn)題發(fā)出483缺陷項(xiàng):研究負(fù)責(zé)人未及時(shí)確認(rèn)或記錄偏離�����;未評(píng)估偏離對(duì)研究完整性的影響����;偏離頻發(fā)但無(wú)根本原因分析。
關(guān)鍵原則:所有偏離必須透明��、及時(shí)��、可追溯����、有評(píng)估。
引自:《Good Laboratory Practice for Nonclinical Studies》一書