注冊用醫(yī)療器械作為產(chǎn)品成功上市前的“樣板”�����,同時(shí)作為注冊申報(bào)的三要素之一�����,其重要性不言而喻�����。
本文將簡要梳理注冊用醫(yī)療器械所需滿足的基本要求�����。
No.1 注冊用醫(yī)療器械與上市后醫(yī)療器械的區(qū)別
1). 標(biāo)簽上沒有注冊證號和生產(chǎn)許可證號��;
2). 需完成比上市后產(chǎn)品更嚴(yán)格的測試與研究��;
3). 其余質(zhì)量與管理要求與上市后醫(yī)療器械一致���。
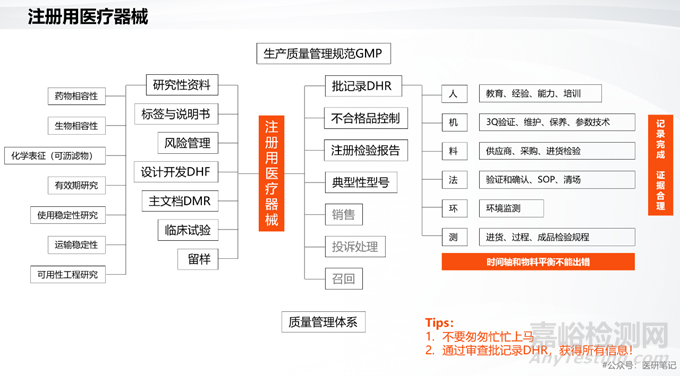
No.2 形成完整的設(shè)計(jì)開發(fā)資料(DHF)
設(shè)計(jì)歷史文檔(DHF)是證明產(chǎn)品設(shè)計(jì)過程受控���、可追溯的核心證據(jù)集。它不僅是一份資料清單��,更是一個(gè)從用戶需求到最終設(shè)計(jì)的完整邏輯閉環(huán)���。
其主要內(nèi)容應(yīng)包括:設(shè)計(jì)開發(fā)策劃��、設(shè)計(jì)開發(fā)輸入���、設(shè)計(jì)開發(fā)輸出、設(shè)計(jì)開發(fā)評審��、審計(jì)開發(fā)驗(yàn)證�����、設(shè)計(jì)開發(fā)變更(若適用)、設(shè)計(jì)開發(fā)確認(rèn)���、設(shè)計(jì)開發(fā)轉(zhuǎn)換。
No.3 醫(yī)療器械的主文檔(DMF)
按照新版的《醫(yī)療器械生產(chǎn)質(zhì)量管理規(guī)范》(GMP)要求和ISO13485的要求�����,必須系統(tǒng)整理并形成醫(yī)療器械主文檔���。
No.4 產(chǎn)品留樣
依據(jù)留樣管理制度進(jìn)行留樣��,并定期開展留樣觀察�����。
No.5 注冊檢測
選擇一款或多款典型性型號進(jìn)行注冊檢驗(yàn)��,其檢驗(yàn)的范圍必須覆蓋同一注冊單元的所有其它型號。
No.6 完成必要的安全和有效性的研究
根據(jù)產(chǎn)品特性開展相關(guān)研究�����,例如:生物相容性�����、藥物相容性、可瀝濾物研究�����、有效期驗(yàn)證�����、使用穩(wěn)定性�����、運(yùn)輸穩(wěn)定性及可用性工程研究等�����。
No.7 開展合規(guī)的臨床評價(jià)
所有第二�����、三類醫(yī)療器械均需通過臨床評價(jià)證明其安全有效性���。主要路徑有三條:免于進(jìn)行臨床評價(jià)�����、通過同品種醫(yī)療器械的臨床數(shù)據(jù)進(jìn)行評價(jià)或開展臨床試驗(yàn)��。
No.8 必須具備完整的批記錄
人員:從事這批生產(chǎn)的人員�����,必須具備相應(yīng)的資質(zhì)要求��,且經(jīng)過必要的培訓(xùn)�����。
設(shè)備:用于該批產(chǎn)品生產(chǎn)的設(shè)備�����、檢驗(yàn)儀器�����、工裝夾具等必須經(jīng)過安裝確認(rèn)��、運(yùn)行確認(rèn)和生產(chǎn)確認(rèn)(3Q)��,且這是設(shè)備的監(jiān)視測量設(shè)備必須經(jīng)過校準(zhǔn)或檢定��。
物料:所有的原材料必須通過經(jīng)過進(jìn)廠檢驗(yàn)��,且供應(yīng)商已被納入合格供方�����。
方法:生產(chǎn)工藝操作規(guī)程�����、檢驗(yàn)的方法��、以及輔助的檢測方法必須文件化��,且經(jīng)過驗(yàn)證或者確認(rèn)�����。
環(huán)境:生產(chǎn)該批醫(yī)療器械的環(huán)境必須符合GMP的要求���,在十萬級��、或者萬級條件下生產(chǎn)�����,并且潔凈車間的環(huán)境經(jīng)過確認(rèn)�����,并且通過第三方的檢測���。
檢測:必須形成進(jìn)貨檢驗(yàn)規(guī)程��、過程檢驗(yàn)規(guī)程�����、成品經(jīng)驗(yàn)規(guī)程���,以及必要的輔助檢驗(yàn)規(guī)定,如環(huán)境檢測��、工藝用水檢測���、工藝用氣檢測等�����。
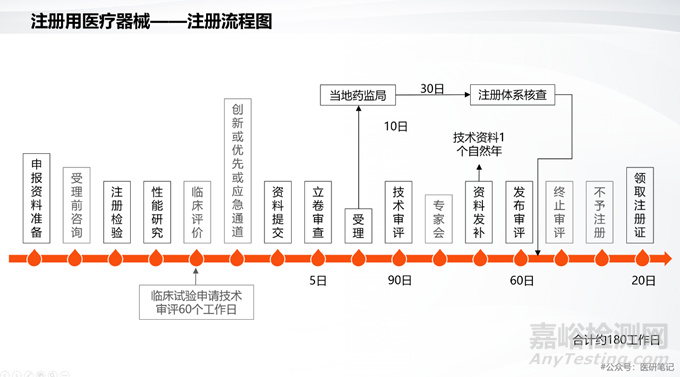
No.9 通過注冊質(zhì)量管理體系核查
注冊申報(bào)過程中�����,還有一個(gè)重要的環(huán)節(jié)——注冊體系現(xiàn)場核查���,旨在確認(rèn)樣品生產(chǎn)過程的真實(shí)性和質(zhì)量管理體系運(yùn)行的有效性。
No.10 輸出注冊申報(bào)資料
依據(jù)《醫(yī)療器械注冊管理辦法》���、注冊申報(bào)資料要求(2021年第121號和122號文)���,輸出注冊申報(bào)資料,為醫(yī)療器械的注冊申報(bào)做準(zhǔn)備���。