為牛骨膠囊用明膠生產(chǎn)企業(yè)提供變更管理的方法,本文從當前國內(nèi)外對藥品生產(chǎn)企業(yè)變更管理的法律法規(guī)要求出發(fā)�,分析了藥品生產(chǎn)企業(yè)變更管理的常見問題。并以某牛骨膠囊用明膠生產(chǎn)企業(yè)依據(jù)《中華人民共和國藥典(2025 年版)》進行的變更管理過程為例���,闡述了變更管理的流程及具體實施步驟�。結果顯示���,該牛骨膠囊用明膠生產(chǎn)企業(yè)嚴格按照相關法律法規(guī)要求建立了變更管理控制系統(tǒng)����,并嚴格實施,降低了牛骨膠囊用明膠產(chǎn)品的質(zhì)量風險�,確保生產(chǎn)出符合相關要求且質(zhì)量可控的牛骨膠囊用明膠。
Part1引言
牛骨膠囊用明膠[1-4]作為藥用輔料����,其產(chǎn)品質(zhì)量的優(yōu)劣直接關乎相關藥品的質(zhì)量,進而對公眾健康及生命安全產(chǎn)生重大影響���。牛骨膠囊用明膠生產(chǎn)企業(yè)只有通過有效的變更管理���,才能降低產(chǎn)品質(zhì)量風險,確保產(chǎn)品的質(zhì)量和安全性����。為此,應對牛骨膠囊用明膠生產(chǎn)企業(yè)如何實施變更管理進行研究 [5]�。
Part2為何開展變更管理
目前�,國內(nèi)外均存在眾多與藥品生產(chǎn)質(zhì)量管理變更控制有關的法律法規(guī)。牛骨膠囊用明膠作為一種藥用輔料���,必須參照并執(zhí)行這些法律法規(guī)的相關要求���。與牛骨膠囊用明膠生產(chǎn)質(zhì)量管理中變更控制有關的法律法規(guī)����,包括國外監(jiān)管機構關于藥品注冊的變更指南�、GMP 標準以及我國關于藥品變更控制的法規(guī)指南等�。
2.1變更管理依據(jù)
2.1.1ICH 指南
《Q12 :藥品生命周期管理的技術和監(jiān)管考慮》為藥品上市后變更管理提供了框架。
2.1.2FDA 藥品生產(chǎn)變更分類管理規(guī)定
根據(jù)藥品安全性�、有效性的潛在影響,將變更分為三大類�。其中,重大變更需要牛骨膠囊用明膠生產(chǎn)企業(yè)向美國食品和藥品監(jiān)督管理局(FDA)提交申請���,并在獲得 FDA 批準后才能實施�。中等變更指在提交變更文件后����,無需等待 FDA 的審批或反饋即可直接實施的變更。而微小變更則無需向 FDA 提交申請或變更文件����,企業(yè)可立即執(zhí)行,僅需在年度報告中進行匯報����。
2.1.3我國法規(guī)指南
我國關于藥品變更控制的法規(guī)指南主要分為生產(chǎn)類變更和注冊類變更兩大類����,并對生產(chǎn)類變更和注冊類變更做了明確規(guī)定 [6]����。
2.2變更控制的常見問題及實施要點
本研究收集整理了國內(nèi)外藥品監(jiān)管或檢查機構對生產(chǎn)企業(yè)提出的近 12 條變更控制相關缺陷,從變更文件體系���、變更評估���、變更分級、變更方案���、變更實施����、變更跟蹤和變更申報與審批等方面對變更控制缺陷進行了分析統(tǒng)計���。其中�,變更文件體系方面的缺陷占比為 27% ����;變更評估方面的缺陷占比為 17% ����;變更分級方面的缺陷占比為 5% ����;變更方案制定不完善方面的缺陷占比為 11% ;變更實施方面的缺陷占比為 31% ���;變更跟蹤方面的缺陷占比為 6% ;變更申報與審批方面的缺陷占比為 3%[6]�。
2.2.1變更文件體系
變更文件體系存在的問題包括:文件體系變更分級不明確;流程圖未涵蓋所有變更控制流程�;缺乏新產(chǎn)品、新設備的變更控制�;未按變更控制程序文件要求開展風險評估工作;發(fā)生重大變更時����,未明確規(guī)定如何與監(jiān)管機構和客戶溝通。
2.2.2變更評估
變更評估方面存在的問題包括:未進行風險評估����;未將變更的所有流程納入整體風險評估�;未對新增品種進行共線風險評估����;未通過評估確定工藝變更后產(chǎn)品是否需要開展穩(wěn)定性考察等。
2.2.3變更分級
變更分級方面的典型缺陷包括變更分級不合理����;變更分級與文件規(guī)定的分級不一致;變更分類不準確���。
2.2.4變更方案
變更方案存在的問題包括:變更方案缺乏實施計劃����,未與監(jiān)管部門溝通報告���;關聯(lián)性變更缺乏詳細的計劃�,部門職責不清���;未明確變更的可接受標準和變更關閉的時機���。
2.2.5變更實施
變更實施存在的問題包括:變更方案批準未能及時啟動;變更物料供應商時未進行工藝驗證或持續(xù)穩(wěn)定性考察�,也未對變更前后輔料的質(zhì)量標準進行對比����;在變更實施過程中���,部分驗證和檢驗數(shù)據(jù)未能記錄���;變更在未獲得監(jiān)管機構批準的情況下即被關閉等。
2.2.6變更跟蹤
變更關閉后���,未對產(chǎn)品的質(zhì)量可比性研究報告和長期穩(wěn)定試驗的實施情況進行跟蹤����,也未進行相應的跟蹤和評估;年度回顧中未對變更進行回顧等�。
2.2.7變更申報與審批
企業(yè)新增生產(chǎn)線時未及時申請變更審批;生產(chǎn)場地的門牌號變更后�,未及時申報、未進行備案且未在年度報告中記錄等�。
Part3如何開展變更管理
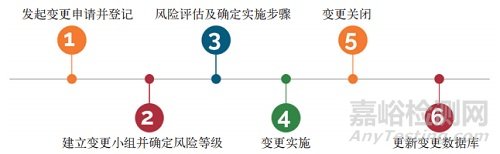
牛骨膠囊用明膠生產(chǎn)企業(yè)根據(jù)相關法律法規(guī)的要求,不僅需要建立變更控制系統(tǒng)����,還需要制定操作規(guī)程���。牛骨膠囊用明膠生產(chǎn)企業(yè)需要根據(jù)已建立的變更控制系統(tǒng)和變更操作規(guī)程對所有變更進行評估����,分析這些變更對牛骨膠囊用明膠產(chǎn)品質(zhì)量的潛在影響����。牛骨膠囊用明膠生產(chǎn)企業(yè)可根據(jù)變更的范圍���、性質(zhì)以及對產(chǎn)品質(zhì)量潛在影響的程度���,對變更進行分類,圖 1 所示為變更管理流程�。
1 變更管理流程
3.1發(fā)起變更申請并登記
若牛骨膠囊用明膠生產(chǎn)企業(yè)需要開展變更,變更所有者需要按照已制定的變更控制程序文件要求�,向質(zhì)量管理部門提出變更申請,并在獲得同意后在該部門進行登記����。例如,某牛骨膠囊用明膠生產(chǎn)企業(yè)在開展 2024 年度管理評審時���,發(fā)現(xiàn)2025 年將實施《中華人民共和國藥典(2025 年版)》�,因此,質(zhì)量管理部門將《中華人民共和國藥典(2025 年版)》變更納入 2025 年變更計劃中���。2025 年 1 月 1 日����,企業(yè) QC 主管就向負責變更管理的 QA 發(fā)起了《中華人民共和國藥典(2025 年版)》變更申請�。負責牛骨膠囊用明膠生產(chǎn)企業(yè)變更管理的 QA 為該變更分配了編號“2025001”����,并在變更項目清單中進行登記。收到 QA 確定的變更編號后�,QC主管按照企業(yè)變更控制程序文件要求����,填寫變更計劃表中的“變更發(fā)起”部分����。具體內(nèi)容包括:變更項目名稱為“《中華人民共和國藥典(2025 年版)》實施”���;變更類型選為“檢驗標準”���;變更過程選為“化驗室”;變更申請日期為“2025/1/1”���;變更編號為“2025001”;變更內(nèi)容描述為“《中華人民共和國藥典(2025 年版)》將于 2025 年 X 月 X日實施�,化驗室需要根據(jù)新版藥典的要求確認現(xiàn)有資源,并根據(jù)確認結果提供所需的資源�,以滿足日常檢驗工作需求”�;變更原因填寫為“《中華人民共和國藥典(2025 年版)》實施,牛骨膠囊用明膠質(zhì)量標準將發(fā)生變更”����。至此����,QC 主管完成變更申請。
3.2建立變更小組并確定風險等級
按照公司變更控制程序文件的要求�,2025 年 1 月 6 日,在公司變更小組管理會議上���,總經(jīng)理指定了《中華人民共和國藥典(2025 年版)》變更管理小組����。變更管理小組按照 1# 變更判斷樹,確定《中華人民共和國藥典(2025年版)》變更的風險等級為 2 級����,即需要通知客戶關于《中華人民共和國藥典(2025 年版)》的事宜。牛骨膠囊用明膠生產(chǎn)企業(yè)因《中華人民共和國藥典(2025 年版)》相關規(guī)定變更�,需通過提交年度報告的方式進行應對。
3.3風險評估及確定實施步驟
按照公司變更控制程序要求���,牛骨膠囊用明膠生產(chǎn)企業(yè)變更管理小組根據(jù)公司變更管理檢查清單對《中華人民共和國藥典(2025 年版)》的變更���,從質(zhì)量、環(huán)境健康安全(EHS)�、工程、生產(chǎn)和維修���、安全以及精益管理等方面進行了風險評估,并根據(jù)評估結果確定變更實施步驟�,明確每步負責人�、所需措施、計劃和實際完成日期和相關記錄。
3.4實施變更
公司銷售經(jīng)理負責通知客戶���,完成變更計劃表中的第二部分“銷售”�。變更管理團隊的其他成員根據(jù)變更管理檢查清單所確定的實施步驟�,逐一執(zhí)行每一步驟所涉及的措施、明確負責人����、設定措施完成截止日期及記錄實際完成日期����,并詳盡記錄每個過程的相關信息,從而完成變更計劃表中的第三部分“項目負責人”相關內(nèi)容的填寫����。項目負責人部分是變更過程的核心。按照以下步驟實施《中華人民共和國藥典(2025 年版)》的變更:
QC 主管申請購買藥典����;
收到《中華人民共和國藥典(2025年版)》后���,QC 主管需核實公司產(chǎn)品與該藥典的相關內(nèi)容相符���,具體需確認《中華人民共和國藥典(2025 年版)四部》中第1016至1017頁的《牛骨膠囊用明膠》標準為公司必須遵循的執(zhí)行標準;
QC 主管要對《中華人民共和國藥典(2025 年版)》和《中華人民共和國藥典(2020 年版)》進行比較���,找出不同之處。再根據(jù)確定的變化點�,確定《中華人民共和國藥典(2025 年版)》換版后,化驗室現(xiàn)有的資源是否足夠�,是否需要增加人員�,檢驗人員是否需要培訓;檢測儀器和設備(包括現(xiàn)有設備的精度)�、檢驗用藥品、試劑和培養(yǎng)基是否需要更新����,檢驗方法是否需要確認和驗證,檢驗作業(yè)指導書和檢驗記錄是否需要增加或者修訂�,產(chǎn)品檢驗報告是否需要修訂。
如果上述工作確定需要變更����,需明確每一個步驟的負責人、措施完成截止日期����、實際完成日期,以及每個過程所形成的記錄���。變更管理檢查清單所列步驟完成后�,變更管理小組需對變更管理檢查清單進行回顧�。如果變更所有項目都已完成,則進行最終確認�。項目完成后,質(zhì)量經(jīng)理�、EHS 經(jīng)理和項目發(fā)起者(QC主管)需在變更管理檢查清單的相應位置填寫各自的姓名和日期,以完成變更管理檢查清單“最終確認”部分���。
3.5關閉變更
牛骨膠囊用明膠生產(chǎn)企業(yè)的變更管理小組在完成變更管理檢查清單中所有要求后���,項目發(fā)起者、質(zhì)量經(jīng)理和 HSE經(jīng)理通過簽名確認變更是否有效�,并填寫日期�,以完成變更計劃表的第四部分“關閉”內(nèi)容?���!吨腥A人民共和國藥典(2025年版)》變更經(jīng)總經(jīng)理指定人員審核���,并由質(zhì)量經(jīng)理批準后正式關閉。
3.6更新數(shù)據(jù)庫
負責變更管理的 QA 人員在獲得已簽字確認的變更管理檢查清單后����,需更新變更項目清單,將 2025 版藥典變更狀態(tài)設置為“已關閉”���。
3.7備案
牛骨膠囊用明膠生產(chǎn)企業(yè)負責藥品年度報告的人員需在《牛骨膠囊用明膠 2025 年度報告》中完成藥品年度報告的填報并提交 [7]�。
Part4結論
牛骨膠囊用明膠生產(chǎn)企業(yè)必須根據(jù)相關法律法規(guī)要求�,建立完善的變更控制系統(tǒng)和變更操作規(guī)程。企業(yè)需要根據(jù)已建立的變更控制系統(tǒng)和變更操作規(guī)程對牛骨膠囊用明膠全生命周期的所有變更進行管理����,確保變更過程受控,從而有效降低牛骨膠囊用明膠產(chǎn)品質(zhì)量風險�,生產(chǎn)出符合注冊和監(jiān)管要求、安全�、有效和質(zhì)量可控的牛骨膠囊用明膠。
參考文獻
[1] 常倩倩���,高斌����,張寧,等 . 淺析膠囊用明膠生產(chǎn)企業(yè)的偏差管理[J]. 流程工業(yè)���,2025(4):70-73.
[2] 陳 世 鵬���, 陳 世會���,董炎超 . 淺析膠囊用明膠持續(xù)性工藝確認[J]. 流程工業(yè)�,2022(1):50-53.
[3] 陳 世 鵬���, 陳 世會 . 牛骨膠囊用明膠瘋牛病的控 制 [J]. 明 膠科 學 與 技 術���,2022(2):98-103.
[4] 王瑩瑩,陳世鵬����,陳世會 . 藥物警戒質(zhì)量管理規(guī)范在明膠生產(chǎn)中的應用[J]. 流程工業(yè),2023(6):16-18.
[5] 國家藥品監(jiān)督管理局 . 國家藥監(jiān)局關于實施 2025 年版《中華人民共和國藥典》有關事宜的公告(2025年 第 32 號)[EB/OL]. (2025-03-25).https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20250325184202175.html.